菲啶衍生物及其制备方法与应用
1.本发明涉及医药领域,具体涉及菲啶衍生物及其制备方法与应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.癌症是当今社会危害人类健康和生命的重大疾病之一。癌症的治疗不仅是医学的一大挑战,而且也导致了严重的社会与经济问题。除手术切除外,大多数患者需要进行保守治疗。近年来,利用癌细胞与正常细胞不同的生化特征实现恶性肿瘤的化学药物治疗备受学者关注。免疫疗法也是一种有前途的治疗方法。虽然对癌症治疗取得了一些进展,但最佳的治疗方法仍不明确,新药的开发仍是必要和紧迫的。
4.现代抗菌化学疗法始于20世纪30年代,在接下来的四十年中,几乎发现了目前使用的所有的抗菌药物的类型。然而,对这些药物的细菌耐药性是目前国际公认的永久性和严重性的问题。因此,发现具有新颖作用方式的药物对于应对耐药性产生的威胁至关重要。基于目前许多抗菌药物的耐药与机体的氧化还原系统有关,靶向机体氧化还原系统发展新作用类型且避免化疗耐药的新抗菌药物至关重要。
技术实现要素:
5.针对现有技术中存在的问题,本发明的目的是提供一种菲啶衍生物及其制备方法与应用,本发明中制备的菲啶衍生物体外对多种癌症细胞株的增殖具有很好的抑制作用,其能抑制硫氧还蛋白还原酶,刺激活性氧积累,诱导氧化应激介导的癌细胞凋亡;体内能明显控制a549异种移植瘤裸鼠模型肿瘤的生长,起到了良好的治疗效果,通过对相关肿瘤标志物的检测也证明了其疗效明显。本发明中制备的菲啶衍生物体外抗菌活性研究表明,本发明公开的菲啶衍生物对于测试的金黄色葡萄球菌表现出很好的生长抑制作用。体内外活性表明这是本发明公开的菲啶衍生物是一种有潜力的治疗癌症、细菌感染的分子,为药物的研发提供一定的基础。
6.为了实现上述目的,本发明的技术方案如下所述:
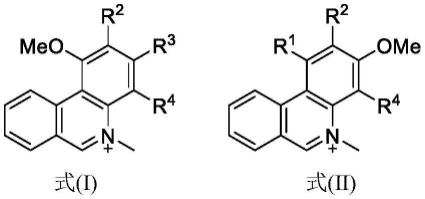
7.在本发明的第一方面,提供一种菲啶衍生物,该衍生物的结构如式x所示:
[0008][0009]
其中,r1选自烷氧基、氢或烷胺基;r2选自氢、氟、甲基、烷氧基、氰基、三氟甲基、正丁基或正戊基;r3选自氢、甲基、烷氧基或氟;r4选自氢、氟、甲基、烷氧基、氰基、三氟甲基、
正丁基或正戊基。
[0010]
在一种或多种实施方式中,所述菲啶衍生物具有式(i)或式(ii)所示结构:
[0011][0012]
优选地,所述r1选自烷氧基;r2选自氢;r3选自烷氧基;r4选自氢。
[0013]
具体地,所述菲啶衍生物选自下列化合物:
[0014]
化合物5c:2-甲基菲啶;
[0015]
化合物5d:2-氟代菲啶;
[0016]
化合物5e:2-甲氧基菲啶;
[0017]
化合物5f:1,3-二甲氧基菲啶;
[0018]
化合物5g:1,2,3-三甲氧基菲啶;
[0019]
化合物6b:5-甲基菲啶-5-碘化物;
[0020]
化合物6c:2,5-二甲基菲啶-5-碘化物;
[0021]
化合物6d:2-氟代-5-甲基菲啶-5-碘化物;
[0022]
化合物6e:2-甲氧基-5-甲基菲啶-5-碘化物;
[0023]
化合物6f:1,3-二甲氧基-5-甲基菲啶-5-碘化物;
[0024]
化合物6g:1,2,3-三甲氧基-5-甲基菲啶-5-碘化物。
[0025]
在本发明的第二方面,提供一种第一方面所述菲啶衍生物的制备方法,包括如下步骤:
[0026]
(1)化合物1在无水thf中,被对甲苯磺酰氯(tscl)磺酰化得到化合物2;随后,化合物2在nai作为催化剂的条件下与2-溴溴苄反应得到化合物3;化合物3与无水k2co3、pd(oac)2和pcy
3-hbf4在氩气保护下加入超干dma反应得到化合物4;
[0027]
(2)化合物4脱ts得到化合物5b-g;
[0028]
(3)化合物5b-g被ch3i甲基化得到化合物6b-g;
[0029]
[0030]
试剂及条件:(a)tscl,吡啶,无水thf;(b)2-溴苄基溴,k2co3,丙酮,回流;(c)pd(oac)2,k2co3,pcy
3-hbf4,无水dma,密封管,130℃,16h;(d)钠/萘,四氢呋喃,-78℃;(e)碘甲烷,甲苯。
[0031]
具体地,所述制备方法如下:
[0032]
(1)将化合物1溶解在无水thf中,加入无水吡啶并搅拌。然后在冰浴下加入对甲苯磺酰氯,室温下搅拌过夜。通过tlc监测反应并在完成后用饱和nahco3溶液淬灭反应。混合物用乙酸乙酯萃取,有机相经合并后用无水na2so4干燥并减压浓缩。混合物经硅胶柱层析纯化(石油醚/乙酸乙酯=1/1),得到化合物2;
[0033]
将化合物2溶解在丙酮中,加入2-溴溴苄和无水k2co3,随后加入催化剂nai,回流,反应冷却后直接减压蒸馏。混合物经硅胶柱层析纯化,得到化合物3;
[0034]
将化合物3、无水k2co3、pd(oac)2和pcy
3-hbf4(44mg,0.12mmol),在氩气保护下加入超干dma,反应加热至130℃反应16~20h。冷却至室温后,用乙酸乙酯萃取,有机相合并后用无水na2so4干燥并减压浓缩。混合物经硅胶柱层析纯化(石油醚/乙酸乙酯=5/1),得到化合物4;
[0035]
(2)将化合物4溶解在20ml无水thf中,并在-78℃下加入到新制备的溶解在无水thf中的na/萘中。搅拌2~4h后,向溶液中通入氧气。将反应置于室温并继续搅拌20~24h。用饱和氯化铵溶液淬灭反应,然后用乙酸乙酯萃取。有机相合并后用无水na2so4干燥并减压浓缩。混合物通过硅胶柱色谱纯化,得到化合物5b-5g。
[0036]
(3)将化合物5b-5g溶解在无水甲苯中,然后加入ch3i,将反应加热至120℃反应4~6h。冷却至室温后,过滤,用甲苯和乙醚洗涤沉淀,收集得到菲啶衍生物。
[0037]
优选地,步骤(2)中所述在-78℃下将化合物4的thf溶液加入到新制备的溶解在无水thf中的na/萘中。
[0038]
在本发明的第三方面,提供了一种药物组合物,其包含上述菲啶衍生物。
[0039]
在本发明的第四方面,提供了一种药物制剂,其包含上述菲啶衍生物或上述药物组合物,以及药学上可接受的载体和/或辅料。所述制剂选自口服制剂和肠胃外给药制剂,可以为片剂、丸剂、胶囊或注射剂。
[0040]
在本发明的第五方面,提供上所述菲啶衍生物或其药学上可接受的盐、组合物在制备抗肿瘤药物中的应用。
[0041]
在一种或多种实施方式中,所述癌症包括人肺癌、宫颈癌、肝癌。
[0042]
体外抗肿瘤活性研究表明,本发明公开的菲啶衍生物对于测试的三种癌细胞a549、hela和hep g2细胞株的增殖具有很好的抑制作用,其能选择性地抑制硫氧还蛋白还原酶,刺激活性氧积累,诱导氧化应激介导的癌细胞凋亡。
[0043]
体内活性研究表明,本发明公开的菲啶衍生物在口服6mg/kg的剂量能明显控制a549异种移植瘤裸鼠模型肿瘤的生长,起到了良好的治疗效果,通过对相关肿瘤标志物的检测也证明了其疗效明显。
[0044]
在本发明的第六方面,提供上所述菲啶衍生物或其药学上可接受的盐、组合物在制备抗菌药物中的应用。
[0045]
体外抗菌活性研究表明,本发明公开的菲啶衍生物对于测试的金黄色葡萄球菌(staphylococcus aureus)表现出很好的生长抑制作用,菲啶衍生物作用24h后的金葡菌的
生长曲线通过测量600nm处的吸光度值,发现不同浓度的菲啶衍生物对金黄色葡萄球菌的生长产生抑制作用。菲啶衍生物通过靶向细菌硫氧还蛋白还原酶对金黄色葡萄球菌的起到抗菌作用,对菲啶衍生物作用的金黄色葡萄球菌提取物进行采用dtnb法进行测定硫氧还蛋白还原酶的活性,发现菲啶衍生物能很好地抑制细菌中的硫氧还蛋白还原酶活性。
[0046]
体内外活性表明这是本发明公开的菲啶衍生物是一种有潜力的治疗癌症、细菌感染的分子,为药物的研发提供一定的基础。
[0047]
如本文所述的,术语“药物组合物”,其还可以是指“组合物”,其可用于在受试者特别是哺乳动物中实现治疗或预防本发明所述疾病或病症。
[0048]
本发明化合物的药物组合物,可以按照以下的任意方式施与:口服、喷雾吸入、鼻腔给药、直肠给药、局部给药、阴道给药、非肠道给药如皮下、肌内、静脉、腹膜内、心室内、鞘内、胸骨内或颅内注射或输入,或借助一种外植的储器用药,其中优选口服、肌注、腹膜内或静脉内用药方式。
[0049]
本发明化合物或含有它的药物组合物可以单位剂量形式给药。给药剂型可以是液体剂型、固体剂型。液体剂型可以是真溶液类、胶体类、乳剂剂型、微粒剂型、混旋剂型。其他剂型例如片剂、滴丸、胶囊、丸剂、气雾剂、粉剂、混悬剂、溶液剂、乳剂、栓剂、颗粒剂、冻干粉针剂、填埋剂、包合物、贴剂、擦剂等。
[0050]
本发明的药物组合物中还可以含有常用的载体,这里所述可药用载体包括但不局限于:离子交换剂,硬脂酸铝,氧化铝,卵磷脂,血清蛋白如人血清蛋白,缓冲物质如磷酸盐,山梨酯,甘油,山梨酸钾,饱和植物脂肪酸的部分甘油酯混合物,水,盐或电解质,如硫酸鱼精蛋白,磷酸氢钾,磷酸氢二钠,氯化钠,锌盐,胶态氧化硅,聚乙烯吡咯烷酮,三硅酸镁,纤维素物质,聚乙二醇,聚丙烯酸酯,羧甲基纤维素钠,蜂蜡,羊毛酯等。载体在药物组合物中的含量可以是1重量%-98重量%,通常大约占到80重量%。为方便起见,局部麻醉剂,防腐剂,缓冲剂等可直接溶于载体中。
[0051]
口服片剂和胶囊可以含有赋形剂如粘合剂,如糖浆,阿拉伯胶,黄芪胶,山梨醇,或聚乙烯吡咯烷酮,填充剂,如乳糖,玉米淀粉,蔗糖,磷酸钙,山梨醇,氨基乙酸,润滑剂,如硬脂酸镁,滑石,聚乙二醇,硅土,崩解剂,如马铃薯淀粉,或可接受的增润剂,如月桂醇钠硫酸盐。片剂可以用制药学上公知的方法包衣。
[0052]
口服液可以制成水和油的悬浮液,溶液,乳浊液,糖浆,也可以制成干品,用前补充水或其它合适的媒质。这种液体制剂可以包含常规的添加剂,如悬浮剂,山梨醇,纤维素甲醚,葡萄糖糖浆,凝胶,羟乙基纤维素,羧甲基纤维素,硬脂酸铝凝胶,氢化的食用油脂,乳化剂,如卵磷脂,山梨聚糖单油酸盐,阿拉伯树胶;或非水载体(可能包含可食用油),如杏仁油,油脂如甘油,乙二醇,或乙醇;防腐剂,如对羟基苯甲酸甲酯或丙酯,山梨酸。如需要可添加调味剂或着色剂。
[0053]
栓剂可包含常规的栓剂基质,如可可黄油或其它甘油酯。
[0054]
对于胃肠外投药,液态剂型通常由化合物和一种消毒的载体制成。载体首选水。依照所选载体和药物浓度的不同,化合物既可溶于载体中也可制成悬浮溶液,在制成注射用溶液时先将化合物溶于水中,过滤消毒后装入封口瓶或安瓿中。
[0055]
必须认识到,通式x、通式(i)~(ii)化合物的最佳给药剂量和间隔是由化合物性质和诸如给药的形式、路径以及所治疗的特定哺乳动物等外部条件决定的,而这一最佳给
药剂量可用常规的技术确定。同时也必须认识到,最佳的疗程,即同时x化合物在额定的时间内每日的剂量,可用本领域内公知的方法确定。
[0056]
本发明的具体实施方式具有以下有益效果:
[0057]
对本发明所公开的菲啶衍生物的体外活性研究表明,其对于测试的三种癌细胞a549、hela和hep g2细胞株的增殖具有很好的抑制作用,其能选择性地抑制硫氧还蛋白还原酶,刺激活性氧积累,诱导氧化应激介导的癌细胞凋亡。
[0058]
对本发明所公开的菲啶衍生物的体内活性研究表明,其在口服6mg/kg的剂量能明显控制a549异种移植瘤裸鼠模型肿瘤的生长,起到了良好的治疗效果,通过对相关肿瘤标志物的检测也证明了其疗效明显;
[0059]
对本发明所公开的菲啶衍生物的体外活性研究表明,其对于测试的金黄色葡萄球菌(staphylococcus aureus)表现出很好的生长抑制作用,发现不同浓度的菲啶衍生物对金黄色葡萄球菌的生长产生抑制作用。菲啶衍生物通过靶向细菌硫氧还蛋白还原酶对金黄色葡萄球菌的起到抗菌作用,对菲啶衍生物作用的金黄色葡萄球菌提取物进行采用dtnb法进行测定硫氧还蛋白还原酶的活性,发现菲啶衍生物能很好地抑制细菌中的硫氧还蛋白还原酶活性。
[0060]
体内外活性表明这是本发明公开的菲啶衍生物是一种有潜力的治疗癌症、细菌感染的分子,为药物的研发提供一定的基础。
附图说明
[0061]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0062]
图1为:6f有效抑制细胞内trxr活性。与对照组相比,被6f处理后a549细胞中trxr的活性呈浓度依赖性抑制。6f处理a549细胞24h后,收集细胞,裂解定量,用胰岛素终点法测细胞内trxr活性。**p《0.01,与对照组对比。
[0063]
图2为:6f有效刺激细胞内活性氧产生。与dmso处理后的细胞比较,被6f处理后a549细胞中活性氧呈浓度依赖性增加。(a)使用dcfh-da对a549细胞进行成像;(b)通过imagej软件测量(a)中的阳性细胞,进行定量。
[0064]
图3为:为6f诱导细胞氧化应激。(a)和(b)6f处理后细胞内gssg水平和gsh/gssg比值。采用酶回收法测定经6f处理24h的a549细胞的胞内gsh和gssg。(c)6f处理后的细胞内总硫醇水平。dtnb滴定用于测定用6f处理24h的a549细胞中的细胞内硫醇。(d)nac或bso对6f细胞毒性的影响。mtt测定用于评估a549细胞与bso或nac孵育24h,然后再孵6f 24h的细胞活力。数据显示为平均值
±
sd,**和^^,p《0.01。
[0065]
图4为:6f诱导a549细胞凋亡。(a)用6f孵育的a549细胞的核形态变化。a549细胞与6f一起孵育24h,然后使用hoechst 33342对细胞核染色30min。比例尺:20μm。(b)经6f处理的a549细胞中caspase3活化的增加。使用比色测定法评估经6f处理24h的a549细胞中的细胞内caspase 3活性。(c)a549细胞的凋亡通过流式细胞术测定。使用annexin v-fitc和pi对用6f处理24h的a549细胞进行染色。细胞数量(q3,活;q2和q4,凋亡;q1,坏死)显示在(d)中。(e)凋亡相关蛋白的表达水平。western印迹用于评估用6f处理24h的a549细胞中的蛋白质。(f)和(g)分别测量(e)中的bax/bcl-2和caspase3/cleaved-caspase 3。数据显示为平
均值
±
sd。*,p《0.05;**和^^,p《0.01。
[0066]
图5为:6f能有效抑制异种移植瘤模型裸鼠的肿瘤生长。(a)a549皮下肿瘤的6f试验设计方案。(b)接种有或没有6f处理的a549细胞的小鼠肿瘤的代表性图像。(c)用6f(0、3和6mg/kg)腹膜内(ip)治疗后异种移植物的肿瘤生长曲线,每种治疗方案持续21天。(d)治疗期间小鼠的体重。(e)每个小鼠肿瘤的重量。(f)裸鼠器官指数无显著差异。数据显示为平均值
±
sd。**,p《0.01对比载体处理组。
[0067]
图6为:6f能有效抑制金黄色葡萄球菌的生长。6f对金黄色葡萄球菌(staphylococcus aureus)作用24h后的生长曲线图。
具体实施方式
[0068]
应该指出,以下详细说明都是例示性的,旨在对本技术提供进一步的说明。除非另有指明,本技术使用的所有技术和科学术语具有与本技术所属技术领域的普通技术人员通常理解的相同含义。
[0069]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本技术的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0070]
下面结合具体的实施例对本发明作进一步的解释和说明。
[0071]
实施例1:1,2,3-三甲氧基-5-甲基菲啶-5-碘化物(6g)
[0072][0073]
试剂及条件:(a)tscl,吡啶,无水thf;(b)2-溴苄基溴,k2co3,丙酮,回流;(c)pd(oac)2,k2co3,pcy
3-hbf4,无水dma,密封管,130℃,16h;(d)钠/萘,四氢呋喃,-78℃;(e)碘甲烷,甲苯。
[0074]
步骤1:4-甲基-n-(3,4,5-三甲氧基苯基)苯磺酰胺(2)的合成
[0075]
将化合物1溶解在无水thf中,加入无水吡啶并搅拌。然后在冰浴下加入对甲苯磺酰氯,室温下搅拌过夜。通过tlc监测反应并在完成后用饱和nahco3溶液淬灭反应。混合物用乙酸乙酯萃取,有机相经合并后用无水na2so4干燥并减压浓缩。混合物经硅胶柱层析纯化(石油醚/乙酸乙酯=1/1),得到化合物2。
[0076]
步骤2:n-(2-溴苄基)-4-甲基-n-(3,4,5-三甲氧基苯基)苯磺酰胺(3)的合成
[0077]
将化合物2溶解在丙酮中,加入2-溴溴苄和无水k2co3,随后加入催化剂nai,回流,
反应冷却后直接减压蒸馏。混合物经硅胶柱层析纯化,得到化合物3。
[0078]
步骤3:1,2,3-三甲氧基-5-甲苯磺酰基-5,6-二氢菲啶(4)的合成
[0079]
将化合物3、无水k2co3、pd(oac)2和pcy
3-hbf4(44mg,0.12mmol),在氩气保护下加入超干dma,反应加热至130℃反应16~20h。冷却至室温后,用乙酸乙酯萃取,有机相合并后用无水na2so4干燥并减压浓缩。混合物经硅胶柱层析纯化(石油醚/乙酸乙酯=5/1),得到化合物4。
[0080]
步骤4:1,2,3-三甲氧基菲啶(5g)的合成
[0081]
将化合物4溶解在20ml无水thf中,并在-78℃下加入到新制备的溶解在无水thf中的na/萘中。搅拌2~4h后,向溶液中通入氧气。将反应置于室温并继续搅拌20~24h。用饱和氯化铵溶液淬灭反应,然后用乙酸乙酯萃取。有机相合并后用无水na2so4干燥并减压浓缩。混合物通过硅胶柱色谱纯化,得到化合物5g。1h nmr(400mhz,cdcl3)δ9.35(dd,j=8.8,0.8hz,1h),9.16(s,1h),8.00(dd,j=8.0,1.6hz,1h),7.85
–
7.80(m,1h),7.66
–
7.62(m,1h),7.49(s,1h),4.06
–
4.04(m,9h).
13
c nmr(100mhz,cdcl3)δ153.91,153.39,151.36,142.75,142.61,132.12,131.20,128.74,126.35,126.15,125.95,112.85,107.09,61.30,60.44,56.04;ms-esi m/z:270.1[m+h]
+
;hplc purity:99.9%(tr=9.540min)。
[0082]
步骤5:1,2,3-三甲氧基-5-甲基菲啶-5-碘化物(6g)的合成
[0083]
将化合物5g溶解在无水甲苯中,然后加入ch3i,将反应加热至120℃反应4~6h。冷却至室温后,过滤,用甲苯和乙醚洗涤沉淀,收集得到6g。黄绿色固体,产率70%。1h nmr(400mhz,dmso-d6)δ10.20(s,1h),9.46(d,j=8.8hz,1h),8.51(dd,j=8.0,1.6hz,1h),8.35
–
8.31(m,1h),8.05
–
8.01(m,1h),7.69(s,1h),4.65(s,3h),4.17
–
4.00(m,9h).
13
c nmr(100mhz,dmso-d6)δ156.23,155.21,152.06,144.75,138.44,133.95,133.16,132.50,129.44,126.22,123.53,115.12,98.63,61.67,61.61,57.51,47.04;ms-esi m/z:284.1[m
–
i]
+
;hplc purity:95.0%(tr=8.176min)。
[0084]
按照与实施例1相同的方法,采用不同的原料,合成以下化合物。
[0085]
实施例2:5-甲基菲啶-5-碘化物(6b)
[0086][0087]
棕色固体,产率78%。1h nmr(400mhz,dmso-d6)δ10.30(s,1h),9.21
–
9.15(m,2h),8.59
–
8.55(m,2h),8.43
–
8.39(m,1h),8.20
–
8.10(m,3h),4.68(s,3h).
13
c nmr(100mhz,dmso-d6)δ156.34,138.29,134.66,134.60,133.03,132.42,130.82,125.83,125.15,124.01,123.65,120.54,46.39;ms-esi m/z:194.1[m
–
i]
+
;hplc purity:99.0%(tr=4.481min).
[0088]
实施例3:2,5-二甲基菲啶-5-碘化物(6c)
[0089][0090]
淡黄色固体,产率75%。1h nmr(400mhz,dmso-d6)δ10.23(s,1h),9.12(d,j=8.4hz,1h),9.00(s,1h),8.54(dd,j=8.0,1.2hz,1h),8.44(d,j=8.8hz,1h),8.40
–
8.36(m,1h),8.09(t,j=7.6hz,1h),8.00(dd,j=8.8,2.0hz,1h),4.65(s,3h),2.70(s,3h).
13
c nmr(100mhz,dmso-d6)δ155.23,141.38,138.02,134.40,133.79,132.90,130.71,125.85,124.51,124.12,123.64,120.29,46.30,21.64;ms-esi m/z:208.1[m
–
i]
+
;hplc purity:98.4%(tr=6.148min).
[0091]
实施例4:2-氟代-5-甲基菲啶-5-碘化物(6d)
[0092][0093]
黄色固体,产率71%。1h nmr(400mhz,dmso-d6)δ10.33(s,1h),9.11(d,j=8.4hz,1h),9.04(dd,j=10.0,3.2hz,1h),8.63(dd,j=9.6,4.8hz,1h),8.57(dd,j=8.0,1.2hz,1h),8.41
–
8.37(m,1h),8.16
–
8.07(m,2h),4.68(s,3h).
13
c nmr(100mhz,dmso-d6)δ162.58(d,j=249.0hz),155.97,138.21,134.17(d,j=4.0hz),133.06,131.45,130.83,128.19(d,j=10.0hz),124.25,124.23,123.88(d,j=9.0hz),120.96(d,j=25.0hz),110.70(d,j=24.0hz),46.74;ms-esi m/z:212.1[m
–
i]
+
;hplc purity:98.8%(tr=4.331min).
[0094]
实施例5:2-甲氧基-5-甲基菲啶-5-碘化物(6e)
[0095][0096]
淡黄色固体,产率72%。1h nmr(400mhz,dmso-d6)δ10.11(s,1h),9.20(d,j=8.4hz,1h),8.53
–
8.46(m,3h),8.36(t,j=8.0hz,1h),8.10(t,j=7.6hz,1h),7.76(dd,j=9.6,2.8hz,1h),4.65(s,3h),4.10(s,3h).
13
c nmr(100mhz,dmso-d6)δ160.77,153.16,137.54,134.06,132.66,130.88,129.42,127.99,124.24,124.16,122.32,122.06,105.92,57.07,46.44;ms-esi m/z:224.1[m
–
i]
+
;hplc purity:99.3%(tr=5.634min).
[0097]
实施例6:1,3-二甲氧基-5-甲基菲啶-5-碘化物(6f)
[0098]
[0099]
黄色固体,产率76%。1h nmr(400mhz,dmso-d6)δ10.21(s,1h),9.48(d,j=8.4hz,1h),8.47(dd,j=8.0,1.6hz,1h),8.31
–
8.26(m,1h),7.99
–
7.95(m,1h),7.42(d,j=2.4hz,1h),7.30(d,j=2.4hz,1h),4.59(s,3h),4.22(s,3h),4.11(s,3h).
13
c nmr(100mhz,dmso-d6)δ162.32,160.33,156.48,138.37,137.70,134.42,133.06,128.74,126.91,123.03,110.98,102.07,94.92,57.56,57.04;ms-esi m/z:254.1[m
–
i]
+
;hplc purity:99.2%(tr=9.295min).
[0100]
实施例7:2-甲基菲啶(5c)
[0101][0102]
橘黄色固体,产率63%。1h nmr(400mhz,cdcl3)δ9.21(s,1h),8.56(d,j=8.0hz,1h),8.33
–
8.32(m,1h),8.08(d,j=8.4hz,1h),8.00(dd,j=8.0,1.2hz,1h),7.84
–
7.80(m,1h),7.69-7.65(m,1h),7.56(dd,j=8.4,2.0hz,1h),2.62(s,3h).
13
c nmr(100mhz,cdcl3)δ152.63,142.75,136.99,132.29,130.76,130.39,129.81,128.69,127.31,126.45,123.92,121.82,21.98;ms-esi m/z:194.1[m+h]
+
;hplc purity:99.5%(tr=7.663min).
[0103]
实施例8:2-氟代菲啶(5d)
[0104][0105]
淡黄色固体,产率67%。1h nmr(400mhz,cdcl3)δ9.21(s,1h),8.43(d,j=8.4hz,1h),8.18
–
8.11(m,2h),8.02(dd,j=8.0,1.2hz,1h),7.86
–
7.82(m,1h),7.74
–
7.69(m,1h),7.49
–
7.44(m,1h).
13
c nmr(100mhz,cdcl3)δ161.24(d,j=246.0hz),152.70,141.17(d,j=2.0hz),132.22(d,j=10.0hz),131.88(d,j=4.0hz),130.96,128.70,128.05,126.23,125.38(d,j=9.0hz),121.95,117.48(d,j=24.0hz),107.08(d,j=24.0hz);ms-esi m/z:198.1[m+h]
+
;hplc purity:95.4%(tr=7.822min).
[0106]
实施例9:2-甲氧基菲啶(5e)
[0107][0108]
白色固体,产率64%。1h nmr(400mhz,cdcl3)δ9.17(d,j=0.4hz,1h),8.55(d,j=8.0hz,1h),8.12(d,j=8.8hz,1h),8.04(dt,j=8.0,0.8hz,1h),7.92(d,j=2.8hz,1h),7.87
–
7.83(m,1h),7.74
–
7.70(m,1h),7.39(dd,j=8.8,2.8hz,1h),4.03(s,3h).
13
c nmr(100mhz,cdcl3)δ158.33,150.92,139.62,131.88,131.34,130.34,128.51,127.35,126.36,125.03,121.71,118.39,102.88,55.50;ms-esi m/z:210.1[m+h]
+
;hplc purity:99.9%(tr=6.793min).
[0109]
实施例10:1,3-二甲氧基菲啶(5f)
[0110][0111]
淡黄色固体,产率69%。1h nmr(400mhz,cdcl3)δ9.35(dd,j=8.4,1.2hz,1h),9.20(s,1h),7.98(dd,j=8.0,1.2hz,1h),7.80
–
7.76(m,1h),7.61
–
7.57(m,1h),7.26(t,j=2.0hz,1h),6.77(d,j=2.4hz,1h),4.08(s,3h),3.97(s,3h).
13
c nmr(100mhz,cdcl3)δ159.67,159.21,154.69,147.75,132.64,131.03,128.59,126.84,126.05,125.75,109.61,102.83,99.49,55.82,55.59;ms-esi m/z:240.1[m+h]
+
;hplc purity:99.8%(tr=8.434min).
[0112]
实施例11:1,2,3-三甲氧基菲啶(5g)
[0113][0114]
淡黄色固体,产率71%。1h nmr(400mhz,cdcl3)δ9.35(dd,j=8.8,0.8hz,1h),9.16(s,1h),8.00(dd,j=8.0,1.6hz,1h),7.85
–
7.80(m,1h),7.66
–
7.62(m,1h),7.49(s,1h),4.06
–
4.04(m,9h).
13
c nmr(100mhz,cdcl3)δ153.91,153.39,151.36,142.75,142.61,132.12,131.20,128.74,126.35,126.15,125.95,112.85,107.09,61.30,60.44,56.04;ms-esi m/z:270.1[m+h]
+
;hplc purity:99.9%(tr=9.540min).
[0115]
实施例12:化合物抑制hela,hepg2和a549细胞增殖活性测定
[0116]
本实施例用于测定本发明菲啶衍生物对常见癌症细胞系hela,hepg2和a549细胞增殖的抑制活性。试验方案如下:将细胞以5000个/孔的浓度接种于96孔透明板中,每孔100μl。在5%co2,37℃条件下培养12h。然后,将无血清培养基中制备的一系列化合物溶液(100μl)加入孔中,继续培养48h,采用mtt法评价细胞存活率,使用酶标仪测量570nm处的吸光度,计算细胞存活率,最后用graphpad prism 8.0计算ic
50
值。本发明受试化合物抑制肿瘤细胞增殖的ic
50
值见下表。
[0117]
表1细胞毒活性和trxr纯酶抑制效果
[0118][0119][0120]
a dtnb法测定孵育2h的trxr纯酶抑制效果
[0121]
b药物浓度为50μm时trxr的剩余活性
[0122]
结论:本发明中的化合物体外细胞增殖表明,化合物6f具有很好的细胞毒性,优于阳性菲啶类化合物nitidine,为设计新型的药物提供了思路。
[0123]
实施例13:体外抑制硫氧还蛋白还原酶试验
[0124]
本实施例用于测定本发明菲啶衍生物对体外硫氧还蛋白还原酶的抑制活性。试验方案如下:在96孔板中,将不同浓度的菲啶衍生物与被nadph预还原的trxr(80nm)在te缓冲液(50mm tris-hcl,1mm edta,ph 7.4)中37℃下孵育2h(终体积为50μl)。用最大引入量的dmso作为对照组。然后加入50μl含有200μm nadph和2.5mm dtnb的te缓冲液,立即使用酶标仪读取前3分钟内412nm处吸光度的线性增加。trxr的活性表示为k
试验组
/k
对照组
*100%。试验结果见表1。
[0125]
结论:6f对硫氧还蛋白还原酶具有很好的抑制作用,优于阳性菲啶类化合物nitidine,为设计新型的化合物提供了思路。
[0126]
实施例14:衍生物6f对细胞硫氧还蛋白还原酶活性影响的测定
[0127]
本实施例用于测定衍生物6f对细胞硫氧还蛋白还原酶活性的影响。试验方案如下:在100mm培养皿中,用不同浓度的6f孵育a549细胞24h。收集细胞后,用ripa裂解液(50mm tris-hcl ph 7.5、2mm edta、150mm nacl、1mm pmsf和1mm na3vo4、1%tritonx-100、0.1%sds、0.5%脱氧胆酸钠)提取并使用bradford法定量细胞总蛋白。将含有20μg蛋白质的裂解液与含有0.33mm胰岛素、15μm e.coli trx和0.66mm nadph的te缓冲液在37℃下孵育30min
(终体积为50μl)。每孔加入200μl含有1mm dtnb的6m盐酸胍(ph=8.0)在室温下孵育5min,使用酶标仪读取412nm处的吸光度。
[0128]
结论:从图1可以看到与dmso处理后的细胞比较,被6f处理后a549细胞中trxr的活性呈浓度依赖性抑制。
[0129]
实施例15:衍生物6f对细胞活性氧影响的测定
[0130]
本实施例用于测定衍生物6f对细胞活性氧活性的影响。试验方案如下:在12孔板中用6f(终浓度为0、1、2、5μm)处理a549细胞6h。然后更换新鲜的含有10μm dcfh-da的无fbs培养基再避光孵育30min。除去培养基并用pbs洗涤细胞3次后,用荧光显微镜(floid cell imaging station)在绿色通道中进行细胞成像。
[0131]
结论:从图2可以看到与dmso处理后的细胞比较,被6f处理后a549细胞中活性氧呈浓度依赖性增加。
[0132]
实施例16:衍生物6f对细胞氧化应激影响的测定
[0133]
本实施例用于测定衍生物6f对细胞氧化应激的影响。试验方案如下:
[0134]
在100mm培养皿中,用0、1、2、5μm的6f孵育a549细胞24h。收集细胞后,在含有1.0%磺基水杨酸和0.1%triton x-100的蛋白提取液(0.1m磷酸钾缓冲液,5mm edta,ph 7.4)中对细胞进行超声裂解。在4℃ 3000g低温离心10min后,立即将上清液转移至新离心管中,用kpe缓冲液稀释样品至磺基水杨酸浓度为0.5%,然后将其分为两组:总gsh组和gssg组。在96孔板中,总gsh组每孔加入20μl稀释4倍的样品以及60μl kpe缓冲液,gssg组每孔加入20μl样品和60μl含有nem的kpe缓冲液(终浓度为2mm)。将96孔板在37℃孵育0.5h后,gssg组每孔额外加入5μl含有cys的kpe缓冲液(cys孵育浓度为2mm),继续孵育0.5h。用含有0.5%磺基水杨酸的kpe缓冲液配制的gsh标准曲线和gssg标准曲线操作与总gsh组和gssg组一致。然后每孔加入含有1.68mm dtnb的60μl kpe缓冲液,再加入含有0.8mm nadph和3.33u/ml gr的60μl kpe缓冲液。立即使用酶标仪读取最初3分钟内412nm处吸光度的线性增加。使用标准曲线计算总gsh和gssg的含量。用总gsh-2*gssg计算出还原态的gsh含量,然后用还原态gsh/gssg的比值表示试验结果。
[0135]
在100mm培养皿中,用不同浓度的6f孵育a549细胞24h。收集细胞后,用ripa裂解液提取总蛋白并定量,将20μl含有30μg总蛋白质的裂解液或20μl标准cys工作曲线加入到96孔板中。然后加入80μl含有1mm dtnb的6m盐酸胍溶液(ph=8.0)在室温下孵育5min。使用酶标仪读取412nm处的吸光度。
[0136]
在96孔板中种6
×
103个细胞与不同浓度的化合物在37℃下孵育24或48小时,每孔重复3组,终体积为100μl。细胞先用50μm bso或10mm nac处理24h,然后再用不同浓度的6f处理细胞,以最大引入量的dmso作为对照组。孵育结束前4h,每孔加入10μl浓度为5mg/ml的mtt。4h后每孔额外加入100μl三联溶解液(5%异丁醇、10%sds、0.1%hcl),然后将96孔板在37℃下孵育过夜。使用酶标仪测量570nm处的吸光度,计算细胞存活率。
[0137]
结论:从图3可以看出,与dmso处理后的细胞比较,6f孵育后的a549细胞中gssg的含量随6f浓度的增加而显著增加,同时gsh/gssg的比值明显降低。此外,随着6f浓度的增加,a549细胞中的还原性总巯基明显减少。nac和bso也可以影响化合物6f的细胞毒性。综上所述,这些结果表明6f导致细胞内产生氧化应激。
[0138]
实施例17:衍生物6f对细胞凋亡影响的测定
[0139]
本实施例用于测定衍生物6f对细胞凋亡的影响。试验方案如下:
[0140]
在12孔板中用6f(终浓度为0、1、2、5μm)处理a549细胞24h。然后更换新鲜的含有5μg/ml hoechst33342的无fbs培养基再避光孵育30min。除去培养基并用pbs洗涤3次后,用荧光显微镜(floid cell imaging station)在蓝色通道中进行细胞成像。
[0141]
在100mm培养皿中,用不同浓度的6f孵育a549细胞24h。收集细胞后,用ripa裂解液提取蛋白并使用bradford法定量细胞总蛋白。在96孔板中,将含有30μg蛋白质的裂解液与含有10mm dtt、5%甘油、0.2mm ac-devd-pna、2mm edta、0.1%chaps、ph 7.4的50mm hepes一起在37℃孵育3h。使用酶标仪读取405nm处的吸光度。
[0142]
在6孔板中用不同浓度的6f处理a549细胞6h后,用pbs洗涤细胞并收集细胞。用annexin v-fitc/pi双染凋亡检测试剂盒进行细胞染色。用流式细胞仪(bd biosciences,usa)检测活细胞、凋亡细胞和坏死细胞。用cell quest软件(bd biosciences,usa)分析处理数据。
[0143]
用不同浓度的6f处理a549细胞24h后,收集细胞并使用ripa裂解液提取并定量细胞总蛋白。用凝胶电泳(sds-page)分离具有等量细胞总蛋白的样品。将蛋白转移到pvdf膜(merck millipore ltd.tullagreen,carrigtwohill,co.cork ireland)上,用5%脱脂牛奶封闭1h,然后分别用trxr1、caspase 3、gapdh、bax或bcl-2一抗(1:1000稀释)在4℃下孵育过夜。用tbst洗涤3次后,用带有hrp的二抗(1:2000)继续孵育pvdf膜1h。用tbst洗涤二抗3次后,使用化学发光试剂盒(glpbio,montclair,ca,usa)来检测蛋白条带。
[0144]
结论:从图4可以看出,用hoechst 33342染色后,6f处理的a549细胞核的淡蓝色荧光逐渐变亮,形状也由大而均匀变为小而聚集,显示出明显的细胞凋亡特征;a549细胞用6f处理后caspase 3活性呈浓度依赖性增加,并且在6f浓度高时显著增加达到对照组的约8倍;利用v-fitc/pi双染试剂盒测定6f处理后的细胞状态,细胞计数结果显示6f以浓度依赖方式触发a549细胞凋亡;6f以浓度依赖的方式影响凋亡相关蛋白的变化,包括bcl-2的减少、bax的增加以及非活性caspase 3转变为被活化的c-caspase 3(即cleaved-caspase 3)。综上所述,所有结果表明6f诱导a549细胞凋亡。
[0145]
实施例18:衍生物6f对异种移植瘤模型裸鼠的治疗效果测定
[0146]
本实施例用于测定衍生物6f对异种移植瘤模型裸鼠的治疗效果。试验方案如下:将15只6周大(~20g)的雄性balb/c裸鼠分成三组,并在右侧大腿注射5
×
106个a549细胞。当肿瘤大小约为70mm3时,对小鼠进行腹腔给药:对照组(含有2%dmso、40%peg400和5%tween-80的生理盐水)、6f(3mg/kg)和6f(6mg/kg),每2天给药一次共21天。每三天记录一次肿瘤的长度、宽度以及小鼠体重。肿瘤体积(v)计算公式为v=lw2/2(l:肿瘤长度,w:肿瘤宽度)。试验结束后解剖小鼠,采集肿瘤组织和各个器官(心肝脾肺肾)用来计算平均肿瘤重量和器官指标。所有程序均按照试验动物研究机构的试验动物护理和使用指南进行。
[0147]
从图5可以看出6f对异种移植瘤模型有较好的治疗作用。结论:从图5可以看出,化合物在裸鼠体内能有效控制肿瘤体积的增长,且体重没有明显下降,器官指数也没有明显变化,显示出其在治疗剂量下的良好疗效和生物相容性。
[0148]
实施例19:衍生物6f对金黄色葡萄球菌抑制的测定
[0149]
本实施例用于测定衍生物6f对金黄色葡萄球菌抑制的测定。试验方案如下:使金黄色葡萄球菌正常生长(37℃,220rpm),直到通过分光光度计法测定600nm处的od值为0.4
时,将其稀释100倍,在96孔板中,用不同浓度的衍生物6f处理,37℃孵育24小时后,测定600nm处的吸光度值。未生长的浓度点即为该样品mic值;将所有未生长的浓度梯度孔划线分离于固体培养基上并培养,无菌落生长的浓度即为该样品的mbc值。
[0150]
结论:从图6可以看出,衍生物6f对金黄色葡萄球菌(staphylococcus aureus)表现出很好的生长抑制作用。
[0151]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1