一种无机-有机杂化纳米材料及其制备方法和应用与流程
1.本发明属于纳米材料技术领域,本发明涉及一种无机-有机杂化纳米材料及其制备方法和应用。
背景技术:
2.高分子材料,如热塑性树脂,已广泛应用于电子工业、塑编行业和汽车内饰等领域。然而,高分子材料的易燃性也对其在这些领域中的应用形成了限制,目前解决该问题的通常手段即为在高分子材料中添加阻燃剂以降低材料的燃烧性能,进而阻止可能发生的大面积燃烧,提升安全性能。
3.目前,较为常用的阻燃剂包括卤系阻燃剂、无机阻燃剂、磷氮系阻燃剂等,其中,无机阻燃剂包括氢氧化铝、碳纳米材料(如碳纳米管和石墨烯等)、蒙脱土等;但是,该些阻燃剂均不同程度地存在一些问题,例如环保性差、在树脂基体中阻燃效率不高、添加量大等,使得高分子材料的广泛应用存在着一定的安全隐患以及性能弊端。
技术实现要素:
4.本发明的目的是克服现有技术的不足,提供一种新的制备无机-有机杂化纳米材料的方法,该方法制成的无机-有机杂化纳米材料可以作为阻燃剂以少量的添加量实现高效且优异的阻燃,且环保性好。
5.本发明同时还提供了一种上述方法制成的无机-有机杂化纳米材料。
6.本发明同时还提供了一种上述方法制成的无机-有机杂化纳米材料在制备阻燃剂以及阻燃型高分子材料中的应用。
7.为达到上述目的,本发明采用的一种技术方案是:一种无机-有机杂化纳米材料的制备方法,该制备方法包括:使季胺盐改性蒙脱石(可以简称ommt)与式(ⅰ)所示化合物在反应压力为0.2mpa以上的封闭环境以及水中发生水热反应,生成无机-有机杂化纳米材料(可以简称asph-nr);;式(ⅰ)中,r1、r2独立地选自苯基、稠环芳烃基团、羟基,r1、r2不同时为羟基,所述苯基、所述稠环芳烃基团均未被取代,所述稠环芳烃基团仅由碳、氢元素构成;其中,当r1、r2独立地选自苯基、羟基时,m、n分别为0;当r1为稠环芳烃基团时,m为1;当r2为稠环芳烃基团时,n为1;所述反应的反应温度为140-200℃,所述季胺盐改性蒙脱石通过使季胺盐与蒙脱石发生阳离子交换反应获得,所述季胺盐改性蒙脱石的层间距大于所述蒙脱石的层间距。
8.根据本发明的一些优选方面,所述稠环芳烃基团为萘基团、蒽基团或菲基团。
9.在本发明的一些优选实施方式中,所述式(ⅰ)所示化合物为二苯基磷酸()和/或2-萘基磷酸()。
10.根据本发明的一些优选方面,所述季胺盐改性蒙脱石与所述式(ⅰ)所示化合物的投料质量比为1∶1-10。
11.进一步地,所述季胺盐改性蒙脱石与所述式(ⅰ)所示化合物的投料质量比为1∶2-4。
12.根据本发明的一些优选方面,所述反应压力为0.5-5mpa。进一步地,在本发明的一些实施方式中,所述反应压力为1-2mpa。
13.根据本发明的一些优选方面,所述反应温度为150-170℃。进一步地,在本发明的一些实施方式中,所述反应温度为155-165℃。
14.根据本发明的一些优选方面,所述季胺盐为选自十六烷基三甲基溴化铵(ctab)、十六烷基三甲基氯化铵(ctac)、十八烷基三甲基溴化铵(stab)、十八烷基三甲基氯化铵(stac)中的一种或多种的组合。
15.根据本发明的一些优选方面,在制备所述季胺盐改性蒙脱石的过程中,所述季胺盐与所述蒙脱石的投料质量比为1-3∶1。
16.在本发明的一些优选实施方式中,所述无机-有机杂化纳米材料的制备方法包括如下实施方式:使季胺盐与蒙脱石发生阳离子交换反应获得季胺盐改性蒙脱石,加入水,超声处理,加入式(ⅰ)所示化合物,混合后转移至反应釜,将反应釜封闭后在加热环境下保持,反应结束后冷却,过滤,洗涤,干燥,得到白色固体,即为所述无机-有机杂化纳米材料。
17.在本发明的一些优选实施方式中,所述超声处理的时间为0.5-3h。
18.在本发明的一些优选实施方式中,所述水为去离子水。
19.在本发明的一些优选实施方式中,所述反应釜可以采用市售的特氟龙不锈钢高压釜。
20.在本发明的一些优选实施方式中,所述加热环境可以由烘箱提供。
21.在本发明的一些实施方式中,所述洗涤可以依次使用去离子水、乙醇、丙酮和乙醚进行清洗。在本发明的一些优选实施方式中,所述干燥可以在50-70℃下进行,干燥时间可以为5-20h。在本发明的一些优选实施方式中,所述水热反应的时间为5-24h,例如可以为5h、6h、7 h、8h、9h、10h、11h、12h、13h、14h、15h、16h、17h、18h、19h、20h、21h、22h、23h、24h等。
22.根据本发明,所述无机-有机杂化纳米材料为棒状结构,该棒状结构包含通过化学键连接的内层和外层,所述内层包含al-o-p键,所述外层包含si-o-al键。
23.在本发明的一些优选实施方式中,所述无机-有机杂化纳米材料的直径约为40-60nm,所述内层的直径约为20-30nm。
24.本发明提供的又一技术方案:一种上述所述的制备方法制成的无机-有机杂化纳米材料。
25.本发明提供的又一技术方案:一种上述所述的无机-有机杂化纳米材料在制备阻
燃剂中的应用。
26.本发明提供的又一技术方案:一种阻燃剂,该阻燃剂包含上述所述的无机-有机杂化纳米材料。
27.本发明提供的又一技术方案:一种阻燃型高分子材料,其包括树脂基体和阻燃材料,该阻燃材料包含上述所述的无机-有机杂化纳米材料或上述所述的阻燃剂。
28.根据本发明的一些优选方面,以质量百分含量计,所述阻燃材料占所述阻燃型高分子材料的1%-20%。
29.进一步地,以质量百分含量计,所述阻燃材料占所述阻燃型高分子材料的4%-12%。
30.在本发明的一些实施方式中,所述阻燃型高分子材料的制备方法包括:使树脂基体、阻燃材料以及选择性的其他材料进行共混,密炼,挤出,造粒;其中,所述的其他材料可以为抗氧剂等等。
31.在本发明的一些实施方式中,所述树脂基体可以为聚乙烯、聚丙烯、聚乳酸(pla)、热塑性聚氨酯弹性体橡胶(tpu)、乙烯-醋酸乙烯酯共聚物(eva)、聚对苯二甲酸乙二醇酯(pet)、聚苯硫醚(pps)和尼龙(nylon)等等。
32.根据本发明,所述蒙脱石可以商购获得,蒙脱石的晶格由二维层组成,这些层以规则的范德华键组织起来形成堆叠,层间存在例如钠离子、钙离子、镁离子等,因此其具有层间阳离子交换的能力,本发明利用季胺盐可以通过阳离子交换反应插层到蒙脱石的层间的能力,进而以扩大蒙脱石的层间距,为后续水热反应的进行提供了基础。
33.进一步地,本发明以扩大了层间距的季胺盐改性蒙脱石与特定的式(ⅰ)所示化合物为原料,使两者在封闭环境中发生水热反应,在水热反应过程中,扩大了层间距的季胺盐改性蒙脱石能够发生剥离、降解与分解,原有的层状结构几乎消失,季胺盐改性蒙脱石的al-o八面体片在特定的水热环境下分解为纳米构建块体;进而,通过与式(ⅰ)所示化合物的反应(al-oh基团与p-oh基团反应),将这些纳米构建块体重构为具有al-o-p键结构的内层材料,并且由于本发明的式(ⅰ)所示化合物具有苯基或稠环芳烃基团,该些基团上π键之间的π-π堆积作用使内层材料具有相互结合的驱动力,促进了材料的重构与组装;此外,季胺盐改性蒙脱石中的si-o四面体也在特定的水热环境下同样发生分解,与具有al-o-p键结构的内层材料发生反应(al-oh基团与si-oh基团反应),形成al-o-si结构,使得 sio2结构能够包裹在al-o-p键结构的内层材料的外层,最终在π-π堆积作用的驱动下,形成了大致为核壳结构的纳米尺寸的棒状结构,即为本发明无机-有机杂化纳米材料。
34.由于上述技术方案运用,本发明与现有技术相比具有下列优点:本发明基于现有阻燃剂存在的诸如环保性差、在树脂基体中阻燃效率不高、添加量大等缺陷,在大量实验研究中,意外发现,当将经过层间扩大的插层结构的季胺盐改性蒙脱石在特定的压力与温度的水热条件下,尤其是在式(ⅰ)所示化合物的参与下,蒙脱石结构发生了结构的降解与重组组装,最终将二维层状的蒙脱石转变为一维的棒状结构的无机-有机杂化纳米材料,实践表明,该新颖结构以及组成上的改变,赋予了无机-有机杂化纳米材料优异的阻燃特性,并且可以以少量的添加量达到高阻燃特性,经过进一步研究分析机理,认为应是本发明的无机-有机杂化纳米材料高温受热之后可以迁移到高分子材料的表面,并且在高温环境下被重新分解释放,通过释放含磷物质使得式(ⅰ)所示化合物的苯环或
芳香稠环在高温下碳化并在高分子材料表面形成类似于石墨烯结构的保护碳层,尤其是受热分解后的含al、含si的纳米颗粒可以很容易地迁移到高分子材料表面,与类似于石墨烯结构的保护碳层共同构成致密的保护层,起到阻隔氧气功能,实现高效且优异的阻燃,同时本发明无机-有机杂化纳米材料的环保性好、易制备,极大地提升了高分子材料的应用安全性与广泛性。
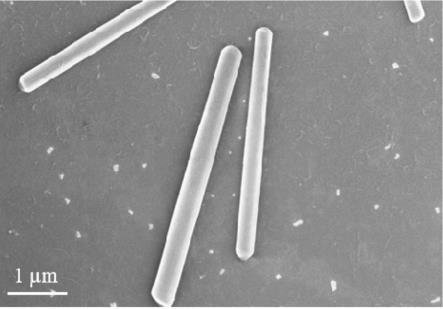
附图说明
35.图1为本发明实施例1制备的无机-有机杂化纳米材料的扫描电镜(sem)图;图2为本发明实施例1制备的无机-有机杂化纳米材料的透射电镜(tem)图之一(标尺为200nm);图3为本发明实施例1制备的无机-有机杂化纳米材料的透射电镜(tem)图之二(标尺为5nm);图4为本发明实施例1制备的无机-有机杂化纳米材料、季胺盐改性蒙脱石以及实施例1制备的无机-有机杂化纳米材料分别在380℃、420℃和450℃处理后的分解残渣的ft-ir光谱谱图;图5为本发明实施例1制备的无机-有机杂化纳米材料、季胺盐改性蒙脱石以及实施例1制备的无机-有机杂化纳米材料分别在380℃、420℃和450℃处理后的分解残渣的xrd谱图;图6为本发明实施例1制备的无机-有机杂化纳米材料在380℃下加热10 min后的分解残渣的扫描电镜(sem)图;图7为在图6处理基础上继续对本发明实施例1制备的无机-有机杂化纳米材料在420℃下加热10 min后的分解残渣的扫描电镜(sem)图;图8为在图7处理基础上继续对本发明实施例1制备的无机-有机杂化纳米材料在450℃下加热10 min后的分解残渣的扫描电镜(sem)图;图9为本发明实施例1、实施例2、实施例3、实施例4、实施例5所制备的无机-有机杂化纳米材料的xrd谱图;图10为本发明应用实施例1:pet/7.8%asph-nr、应用对比例1:pet/7.8% mp、应用对比例2:pet/7.8% ommt经燃烧处理之后的残碳的扫描电镜(sem)图;图11为本发明应用对比例1:pet/7.8% mp经燃烧处理之后的残碳的能谱(eds)图;图12为本发明应用对比例2:pet/7.8% ommt经燃烧处理之后的残碳的能谱(eds)图;图13为本发明应用实施例1:pet/7.8%asph-nr经燃烧处理之后的残碳的能谱(eds)图。
具体实施方式
36.以下结合具体实施例对上述方案做进一步说明;应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
37.下述实施例中未作特殊说明,所有原料均来自于商购或通过本领域的常规方法制备而得。蒙脱石购自南京先丰纳米材料有限公司,牌号xf144;聚对苯二甲酸乙二醇酯(pet)购自美国杜邦公司,牌号re5264;特氟龙不锈钢高压釜为西安洪辰仪器厂,型号20170918。
38.实施例1本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石(ommt)的分散液采用十六烷基三甲基溴化铵(ctab)与蒙脱石(质量比为 3:1),通过发生阳离子交换反应得到ommt,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石(ommt)的分散液;(2)制备无机-有机杂化纳米材料(可以简称asph-nr)将二苯基磷酸(dppa)加入步骤(1)得到的季胺盐改性蒙脱石(ommt)的分散液中,其中ommt与dppa的质量之比为1:3,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
39.对该无机-有机杂化纳米材料进行表征,图1为扫描电镜(sem)图,图2为透射电镜(tem)图之一(标尺为200nm),图3为透射电镜(tem)图之二(标尺为5nm),可以看出制备出的无机-有机杂化纳米材料呈棒状结构,且包含内层和外层。
40.图4本发明实施例1制备的无机-有机杂化纳米材料、季胺盐改性蒙脱石以及实施例1制备的无机-有机杂化纳米材料分别在380℃、420℃和450℃处理后的分解残渣的ft-ir光谱谱图。p-o键对应的特征吸附带明显减少。同时,在1050 cm-1
处出现了al-o键拉伸的吸附带,结果表明al-o-p键在高温下发生断裂,从而释放出含p的物质。
41.图5为本发明实施例1制备的无机-有机杂化纳米材料、季胺盐改性蒙脱石以及实施例1制备的无机-有机杂化纳米材料分别在380℃、420℃和450℃处理后的分解残渣的xrd谱图,xrd结果表明,在7.3
°
时,随着温度的升高,衍射图谱发生了衰减。450℃加热所得残渣的xrd谱图与ommt相似,表明含p的物质在asph-nr结构中被重新分解释放,在结构组成上与ommt基本一致。
42.图4和图5的这些结果也揭示了asph-nr分解主要通过释放含磷物质进而产生硅铝类物质。利用扫描电镜进一步研究了asph-nr的分解过程。图6-8分别为依次经380℃、420℃和450℃加热10 min后的asph-nr的形貌。可以看出,380℃加热后,asph-nr基本保持了原来的一维纳米棒形貌。然而,在420℃处理时,虽然一维纳米结构仍然存在,但出现了项链状形态,表明asph-nr的初步分解。当加热温度提高到450℃时,一维纳米结构消失,而出现了大量纳米尺寸的颗粒。如前所述,这些纳米颗粒主要由硅铝类物质组成,表明受热分解后的al、si的纳米颗粒可以迁移到聚合物表面并形成致密的保护层。
43.实施例2本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十六烷基三甲基溴化铵(ctab)与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石
的分散液;(2)制备无机-有机杂化纳米材料将二苯基磷酸(dppa)加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季胺盐改性蒙脱石与二苯基磷酸的质量之比为1:1,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
44.实施例3本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十六烷基三甲基溴化铵(ctab)与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石的分散液;(2)制备无机-有机杂化纳米材料将二苯基磷酸(dppa)加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季胺盐改性蒙脱石与二苯基磷酸的质量之比为1:2,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
45.实施例4本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十六烷基三甲基溴化铵(ctab)与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石的分散液;(2)制备无机-有机杂化纳米材料将二苯基磷酸(dppa)加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季胺盐改性蒙脱石与二苯基磷酸的质量之比为1:4,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
46.实施例5本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十六烷基三甲基溴化铵(ctab)与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石的分散液;(2)制备无机-有机杂化纳米材料将二苯基磷酸(dppa)加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季
胺盐改性蒙脱石与二苯基磷酸的质量之比为1:5,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
47.将实施例1至实施例5所获的无机-有机杂化纳米材料分别进行xrd表征,具体如图9所示,当季胺盐改性蒙脱石与二苯基磷酸的质量之比从1:1提高到1:3时,在约6
°
特征峰峰值升高。进一步将比值提高到1:5,该峰值出现下降。这个结果表明季胺盐改性蒙脱石与二苯基磷酸的反应为缩聚反应。
48.实施例6本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十六烷基三甲基溴化铵与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石的分散液;(2)制备无机-有机杂化纳米材料将2-萘基磷酸加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季胺盐改性蒙脱石与2-萘基磷酸的质量之比为1:3,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
49.实施例7本例提供一种无机-有机杂化纳米材料的制备方法,该制备方法包括:(1)制备季胺盐改性蒙脱石的分散液采用十八烷基三甲基溴化铵与蒙脱石(质量比为 3:1)通过发生阳离子交换反应得到季胺盐改性蒙脱石,然后加入50 ml去离子水,超声1h,获得季胺盐改性蒙脱石的分散液;(2)制备无机-有机杂化纳米材料将2-萘基磷酸加入步骤(1)得到的季胺盐改性蒙脱石的分散液中,其中季胺盐改性蒙脱石与2-萘基磷酸的质量之比为1:3,搅拌得到混合溶液,将该混合溶液转移到特氟龙不锈钢高压釜后,送入烘箱中160℃恒温保持12 h,将溶液冷却至室温,过滤,用去离子水、乙醇、丙酮和乙醚依次清洗残留物,然后在真空烘箱中60℃干燥12h,得到白色固体,即为无机-有机杂化纳米材料。
50.应用实施例1将实施例1制备的无机-有机杂化纳米材料与聚对苯二甲酸乙二醇酯(pet)通过密炼机共混(密炼温度为:280℃),然后挤出造粒(挤出温度为:加料段:260℃,融化段:280℃,计量段:270℃),干燥,获得阻燃型高分子材料;其中,以质量百分含量计,无机-有机杂化纳米材料的添加量占聚对苯二甲酸乙二醇酯(pet)的添加量的7.8%。因此,本例制成的阻燃型高分子材料可以简称pet/7.8%asph-nr。
51.应用实施例2
基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为实施例2制备的无机-有机杂化纳米材料。
52.应用实施例3基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为实施例3制备的无机-有机杂化纳米材料。
53.应用实施例4基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为实施例4制备的无机-有机杂化纳米材料。
54.应用实施例5基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为实施例5制备的无机-有机杂化纳米材料。
55.应用对比例1基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为三聚氰胺磷酸盐(mp),所述三聚氰胺磷酸盐(mp)是将三聚氰胺与磷酸反应,沉淀过滤后并在真空中干燥制得。本例制成的阻燃型高分子材料可以简称pet/7.8% mp。
56.应用对比例2基本同应用实施例1,其区别仅在于,将实施例1制备的无机-有机杂化纳米材料替换为实施例1中步骤(1)制备的季胺盐改性蒙脱石(ommt)。本例制成的阻燃型高分子材料可以简称pet/7.8% ommt。
57.性能测试将纯的聚对苯二甲酸乙二醇酯(pet)、应用实施例1以及应用对比例1-2所的阻燃型高分子材料进行如下性能测试,具体结果参见表1所示:
。
58.表1表明,在聚酯pet中加入7.8%的asph-nr后,loi大大提高到31.6%,pet/7.8%asph-nr的loi远高于pet/7.8%ommt和pet/7.8%mp。此外,在聚酯中添加7.8wt%的asph-nr可使phrr降低约50%,phrr在658kw
·
m-2
,也低于pet/7.8%ommt和pet/7.8%mp的phrr。虽然pet/7.8%asph-nr的t
ign
较其他材料短,但t
phrr
可延长至230 s。结果表明,asph-nr的加入使pet具有良好的阻燃性能。值得注意的是,与mp和ommt阻燃剂相比,asph-nr在降低pet峰值热释放速率、平均质量损失率和提高极限氧指数方面均具有明显的优势。
59.进一步地,对应用实施例1:pet/7.8%asph-nr、应用对比例1:pet/7.8% mp、应用对比例2:pet/7.8% ommt经燃烧处理之后的残碳分别进行表征,其扫描电镜(sem)图如图10所示,从sem图(上部为标尺是1μm的形态,下部为标尺是200nm的形态)中可以看出,pet/7.8%asph-nr的残炭呈致密结构,而pet/7.8% ommt和pet/7.8% mp的残炭呈高孔隙形态,未形成保护层。分析认为,致密的结构,具有高耐热性,可以使气体和热量隔离,应是pet/7.8%asph-nr具有出色的阻燃性能的原因。sem图像进一步表明,致密的结构应是由大量的超小尺寸颗粒构成的。能谱(eds)图结果表明,这些颗粒由al、p、si、o和c元素组成,分别可能对应氧化铝、聚磷酸盐类、二氧化硅和碳层(类石墨烯结构),结果表明,由asph-nr(能谱(eds)图为图13)分解生成的硅铝类物质的纳米颗粒很容易迁移到聚酯表面,表面的含磷颗粒可能对应焦磷酸盐和聚磷酸盐类型结构。相反,pet/7.8% ommt的能谱(eds)图(图12)中基本没有si和al信号,这可以解释为,由于层状结构,ommt几乎不迁移到表面。在pet/7.8% mp的能谱(eds)图(图11)中仅能观察到微弱的p信号,结果表明,mp在该体系中主要转化为含磷气体,几乎没有半焦例如焦磷酸盐等的生成。
60.上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人
士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
61.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1