一种二维有机无机杂化的多酸基高核稀土衍生物及其制备
1.本发明属于有机无机复合材料技术领域,特别涉及一种二维有机无机杂化的多酸基高核稀土衍生物及其制备和应用。
背景技术:
2.多金属氧酸盐,简称多酸(polyoxometalates,简写为poms),由前过渡金属离子(mo
6+
、w
6+
、v
5+
、nb
5+
、ta
5+
等)通过氧原子桥连而成的无机纳米级金属氧簇(参见m.t.pope,a.m
ü
ller,angew.chem.int.ed.1991,30,34-48)。缺位多酸因其富氧的表面和高负电荷可以很好地结合过渡金属离子和稀土离子聚集成簇,同时其可以作为节点构建多维框架。多酸基高核金属簇合物由于其独特的结构特征和在磁性材料、光学材料以及路易斯酸催化等领域的潜在应用而受到了广泛的关注(参见s.t.zheng,g.y.yang,chem soc rev.2012,41,7623-46;s.s.wang,g.y.yang,chem.rev.2015,115,4893-4962)。
3.目前,多酸基高核过渡金属簇的合成、结构和性质研究已被广泛报道,如{mn
19
(oh)
12
(siw
10o37
)6}中mn
19-簇呈现出有趣的平面结构,co
16-簇{[co4(oh)3po4]4(pw9o
34
)4]}表现出良好的单分子磁体性质,ni
25-簇{ni
25
(h2o)2(oh)
18
(co3)2(po4)6(siw9o
34
)6}具有优异的光催化活性,zr
24-簇{zr
24o22
(oh)
10
(h2o)2(w2o
10
)2(gew9o
34
)4(gew8o
31
)2}可以高效的催化硫醚转化为亚砜(参考b.s.bassil,u.kortz,angew.chem.int.ed.2011,50,5961-5964;m.ibrahim,u.kortz,angew.chem.int.ed.2011,50,4708-4711;x.b.han,e.b.wang,j.am.chem.soc.2015,137,5486-5493;l.huang,g.y.yang,j.am.chem.soc.2014,136,7637-7642)。相比于过渡金属,稀土离子的配位几何和配位数更加的灵活多变,且其正电性和半径更大,使其容易成为多酸单元的桥连离子,从而导致多酸基高核稀土簇的发展较为缓慢,且多集中在纯无机体系yb
6-簇{yb6p4w
30o112
(μ
6-o)(μ
3-oh)6(h2o)6},ce
10-簇{ce
10
p6w
48o183
(oh)6(co3)(h2o)
11
}和ln
26-簇{ln
27
ge
10w106o406
(oh)4(h2o)
24
}(参考x.fang,c.l.hill,chem.eur.j.2005,11,712-718;p.ma,j.wang,inorg.chem.2016,55,918-924;z.li,s.t.zheng,et al,angew.chem.int.ed.2017,56,2664
–
2669)。有机配体的引入不仅可以缓解稀土离子的水解,还可以作为合适的电子供体调控结构和性质,是构建多酸基高核稀土簇的有效策略。
[0004]
但目前利用有机配体构建多酸基高核稀土簇合物的研究相对较少,且现有技术中并未有关于二维有机无机杂化的多酸基高核稀土衍生物的相关报道。
技术实现要素:
[0005]
针对上述缺陷,本发明提供一种有机无机杂化的多酸基高核稀土衍生物,其具有二维结构,所得二维有机无机杂化的多酸基高核稀土衍生物可以作为荧光材料和催化材料应用。
[0006]
本发明的技术方案:
[0007]
本发明要解决的第一个技术问题是提供一种有机无机杂化的多酸基高核稀土衍
生物,其分子式为k2[tb6(ch5no3p)4(h2o)
22
(siw
11o39
)2]
·
23h2o,其为二维结构。
[0008]
本发明要解决的第二个技术问题是提供上述有机无机杂化的多酸基高核稀土衍生物的制备方法,所述制备方法为:将多酸前驱体na
10
[a-α-siw9o
34
]
·
18h2o和有机配体氨甲基膦酸溶解在蒸馏水中于75~85℃搅拌反应1~1.5h;再加入tbcl3·
6h2o搅拌混匀;然后调节ph为2.5~3.5,于75~85℃搅拌下继续反应1~1.5h;冷却至室温,析出的晶体即为二维有机无机杂化的多酸基高核稀土衍生物。
[0009]
进一步,所述有机无机杂化的多酸基高核稀土衍生物的制备方法包括以下步骤:
[0010]
1)将多酸前驱体na
10
[a-α-siw9o
34
]
·
18h2o(缺位keggin型多酸)和氨甲基膦酸溶解在蒸馏水中,75~85℃水浴中搅拌1~1.5h;
[0011]
2)将tbcl3·
6h2o加入到步骤1)的反应溶液中,得到白色浑浊溶液;用稀释盐酸溶液和氢氧化钾溶液调节反应溶液的ph为2.5~3.5,热水浴中搅拌下继续反应1~1.5h;冷却至室温后过滤得到无色块状晶体,即为目标产物。
[0012]
进一步,步骤1)中,所述na
10
[a-α-siw9o
34
]
·
18h2o与氨甲基膦酸的摩尔比为:0.1:0.2~0.1:0.3。
[0013]
进一步,步骤1)中,所述na
10
[a-α-siw9o
34
]
·
18h2o的物质的量与蒸馏水的体积比为:0.1mmol:8ml~0.1mmol:12ml。
[0014]
进一步,步骤2)中,所述na
10
[a-α-siw9o
34
]
·
18h2o与tbcl3·
6h2o的摩尔比为:0.1:0.2~0.1:0.3。
[0015]
进一步,步骤2)中,所述的稀释盐酸溶液和氢氧化钾溶液浓度比为1:1。
[0016]
本发明要解决的第三个技术问题是指出上述有机无机杂化的多酸基高核稀土衍生物作为荧光材料或催化材料的应用。
[0017]
本发明要解决的第四个技术问题是提供一种催化材料,所述催化材料包括上述有机无机杂化的多酸基高核稀土衍生物和导电填料(乙炔黑),有机无机杂化的多酸基高核稀土衍生物和导电填料的质量比为5:1~2:1(优选为3:1)。
[0018]
本发明的有益效果:
[0019]
本发明所得有机无机杂化的多酸基高核稀土衍生物可以作为荧光材料和电催化材料,与传统的荧光材料和电催化材料相比,本发明的二维有机无机杂化的多酸基高核稀土衍生物具有以下优点:
[0020]
1)本发明采用在水溶液中自组装的合成策略,成本较低,制备工艺安全简单,容易实现;
[0021]
2)本发明提供的稀土衍生物是晶态材料,可以利用x射线单晶衍射精确解析出其详细的分子结构;
[0022]
3)本发明提供的稀土衍生物稳定性高,在固态和酸性溶液中均可保持骨架稳定,有利于重复使用;
[0023]
4)本发明提供的稀土衍生物具有荧光性质和电催化氧化还原反应等多功能性。
附图说明:
[0024]
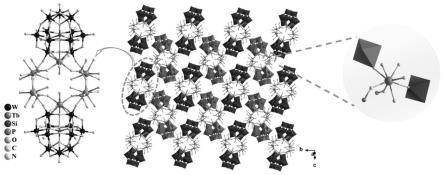
图1是本发明实施例1所得化合物1的多阴离子的多面体示意图及二维结构示意图。
[0025]
图2为本发明化合物1的红外图谱。
[0026]
图3为本发明化合物1的热重图谱。
[0027]
图4为本发明化合物1的实验粉末x射线衍射图谱和模拟图谱对比示意图,证明实验所用样品纯度很高。
[0028]
图5为本发明化合物1的发射光谱(a)和在室温下的寿命衰减曲线及拟合曲线(b)。
[0029]
图6(a)为本发明实施例1所得化合物1在264nm的激发光照射下随时间变化的发射光谱,图(b)为其能量跃迁示意图。
[0030]
图7为本发明实施例1所得化合物1所制备电极材料在0.1mol/l硫酸溶液中不同扫速下的循环伏安曲线(a)和峰电流与扫速的关系示意图(b)(粘附于玻碳电极上)。
[0031]
图8为本发明化合物1所制备电极材料在含亚硝酸根(a)和维生素c(b)的0.1mol/l硫酸溶液中循环伏安曲线随浓度增大的变化趋势。
[0032]
图9为本发明化合物1所制备电极材料在电催化前后的实验粉末x射线衍射图谱,1为反应后图谱,2为反应前图谱,3为模拟图谱。
[0033]
图10为对比例1所得化合物的结构示意图。
[0034]
图11为对比例2所得化合物的结构示意图。
[0035]
图12为对比例3所得化合物的结构示意图。
具体实施方式:
[0036]
本发明提供一种二维有机无机杂化的多酸基高核稀土衍生物,其分子式为k2[tb6(amph)4(h2o)
22
(siw
11o39
)2]
·
23h2o,其中,amph为氨甲基膦酸(ch5no3p)。
[0037]
本发明采用简单的常规水溶液法,利用多酸前驱体原位形成的高电负性的[siw
11o39
]
8-为构筑单元,同时引入酸性的氨甲基膦酸配体可以有效抑制稀土离子的水解并与稀土离子配位,成功制备了以多酸单元为节点的二维有机无机杂化的多酸基高核稀土衍生物的晶态材料。即本发明通过引入强电负性的有机膦酸配体,调控稀土离子的电子结构聚集成簇,同时以si为多酸杂原子调节多酸结构的电荷,改变分子间静电作用力形成二维层状结构。
[0038]
在该稀土衍生物中,[siw
11o39
]
8-单元和氨甲基膦酸配体都能够敏化稀土离子发光,其具有长达43.36μs的荧光寿命。此外,得益于多酸单元所具有的多电子转移的氧化还原特性,该衍生物对不同浓度的维生素c和亚硝酸根具有良好的响应。
[0039]
以下以通过具体实施例对本发明作进一步的详细说明,但本发明的保护范围并不局限于此。
[0040]
实施例1
[0041]
二维有机无机杂化的多酸基高核稀土衍生物采用以下步骤制得:
[0042]
1)按文献报道的方法合成na
10
[a-α-siw9o
34
]
·
18h2o前驱体(参见g.herv
é
,a.t
ézé
*,inorg.chem.1976,27,96-104);
[0043]
2)将0.566g na
10
[a-α-siw9o
34
]
·
18h2o(0.10mmol)和0.033g氨甲基膦酸(0.30mmol)溶解在10ml蒸馏水中,80℃水浴中搅拌下反应1.5h;
[0044]
3)将0.074g tbcl3·
6h2o(0.20mmol)加入到步骤2反应溶液中,得到白色浑浊液;用1mol/l的稀盐酸和1mol/l的氢氧化钾溶液调节溶液ph=3.0
±
0.2,80℃水浴中搅拌下继
续反应1h;反应结束后,将溶液冷却、过滤,滤液静置缓慢挥发一周后析出无色块状晶体,即得目标产物二维有机无机杂化多酸基高核稀土衍生物(记为化合物1)。
[0045]
应用例1
[0046]
本发明化合物1作为电催化材料的具体制备过程如下:
[0047]
1)将化合物1和乙炔黑以3:1的质量比分散在50μl异丙醇、50μl乙醇和50μl萘酚的混合液中;
[0048]
2)将步骤1中混合液超声2h使其分散均匀制成催化材料;
[0049]
3)取出10μl步骤2中的复合材料滴在抛光的玻碳电极上,室温下晾干,再重复滴一次,晾干后就制得了1-gce复合电极。
[0050]
性能测试:
[0051]
采用x-ray单晶衍射技术对上述实施例1制备得到的化合物1进行晶体结构测定和表征,其晶胞参数如下:晶系为单斜,空间群为p21/n;晶胞参数为a=11.9618(2),b=21.9617(4),c=23.5701(5),β=91.919(2),v=6188.4(2)。
[0052]
图1是本发明化合物1的多阴离子的多面体示意图及二维结构示意图;多酸阴离子呈夹心型结构,由两个中心对称的单缺位keggin型[siw
11o39
]
8-单元包夹一个有机无机杂化的tb
6-簇[tb6(amph)4(amph2)(h2o)
23
]
14+
{dy6}核而成;tb
6-簇可以简化为两个共边相连的四面体,其二面角为111.812(4)
°
;tb
6-簇中包含三个晶体学独立的tb离子,tb1和tb3呈现出四方反棱柱的几何构型,而tb2为畸变的双帽三棱柱构型。tb
6-簇中tb1离子由四个来自[siw
11o39
]
8-单元的μ
2-o,两个来自氨甲基膦酸配体的桥氧,和两个端基水完成八配位;tb2/tb3离子配位环境与tb1离子相似,除了三个来自[siw
11o39
]
8-单元的μ
2-o被端基水分子取代,tb-o的键长范围为2.228(8)-2.566(8)。tb3离子作为连接点将相邻的多酸阴离子通过w-o-tb拓展为(4,4)-网格状拓扑结构,对应的w4-o29-tb3和w7-o51-tb3键角分别为144.4(4)
°
和152.1(4)
°
,相邻的多酸阴离子交错排列,其夹角为33.4
°
。
[0053]
图2是本发明化合物1的红外光谱图。其中500-1200cm-1
范围内对应于多酸骨架的特征峰,其中950cm-1
对应于多酸骨架中w
–ot
伸缩振动,在840和780cm-1
处对应于w-o-w的伸缩振动,1160和1400cm-1
处对应于p-o的特征振动带,720-520cm-1
对应于p-c的伸缩振动,3470cm-1
和1627cm-1
分别对应于-oh的伸缩和弯曲振动。
[0054]
图3是本发明化合物1的热重曲线图。化合物1在30-400℃范围内呈现出一步失重过程,失重量为10.64%,对应于20个结晶水和22个配位水的失去(理论值10.61%)。
[0055]
图4是本发明化合物1的实验粉末x射线衍射和模拟x射线衍射(根据单晶衍射数据)对比示意图。实验图谱中衍射峰位置和模拟图谱基本一致,证明化合物1的样品纯度很高,峰强度不同可能是由于化合物1的粉末x射线衍射峰在收集过程中优先取向不同引起的。
[0056]
图5是本发明化合物1的发射光谱图和寿命衰减曲线及拟合曲线。室温下,在378nm的激发光照射下,化合物1的荧光发射光谱在490、544、584与588和620nm出现四个发射峰(a),分别对应于tb
3+
的5d4→7f6,5d4→7f5,5d4→7f4和5d4→7f
3 f-f跃迁。在最强发射(544nm)的条件下,测得化合物1的寿命衰减曲线(b),对其进行单指数函数拟合,寿命为43.36μs,指前因子801.37。
[0057]
图6是本发明化合物1的三维时间分辨光谱和能量转移示意图。室温下,在264nm的
激发光照射下,化合物1的时间分辨光谱证明了从多酸组分到tb
3+
离子能量传递过程(a)。在450nm附近出现宽峰,归因于分子间能量从o
→
m(m=mo or w)传递到tb
3+
离子上,在490和540nm出发射出tb
3+
的离子特征峰,分别归属于tb
3+
离子的5d4→7f6和5d4→7f5跃迁。随着时间的推移,在110μs时峰强度达到最大值,随后逐渐减弱,分别在163、251和95μs时消失。从图中可以看出多酸组分的3t
1u
→1a
1g
跃迁发射峰强度降低很快,说明多酸单元到tb
3+
离子的能量转移过程在一定程度上抑制了tb
3+
中心的发射衰减。基于上述能量转移过程的分析,多酸单元到tb
3+
离子的能量转移过程示意图如图b所示,多酸单元在化合物1中作为敏化剂吸收能量,从1a
1g
基态跃迁到1t
1u
激发态,通过非辐射跃迁过程快速回到3t
1u
激发态,然后从多酸组分的o
→
w lmct光激发3t
1u
三重态通过分子内能量传递过程将能量传递到tb
3+
离子的5d4能级,最终tb
3+
离子在490和540nm位置发射出特征荧光发射峰。
[0058]
图7是本发明化合物1所制备的1-gce复合材料的循环伏安图(a)和峰电流与扫速的关系示意图(b)。在0.1mol/l硫酸溶液和扫描速率为100mv s-1
条件下(a),该cv曲线在-0.8-0.5v范围内出现了3对氧化还原峰,根据氧化还原峰的平均峰电位e
1/2
=(e
pa
+e
pc
)/2公式,它们的e
1/
2分别为-0.318(i-i’)、-0.474(ii-ii’)和-0.636v(iii-iii’)(ag/agcl为参比电极)。扫描速率对1-gce的电化学行为的影响如图b所示,在20到180mv s-1
的扫描速率范围内,电流强度(i
pc
)和扫描速率(v)呈线性关系,表明电化学过程为表面控制过程。
[0059]
图8为本发明化合物1所制备电极材料在含亚硝酸根(a)和维生素c(b)的循环伏安曲线随浓度增大的变化趋势。随着亚硝酸根浓度的增大,还原峰明显增强,同时氧化峰逐渐减弱(a);而随着维生素c浓度的逐渐增加,cv曲线中氧化峰迅速增强(b)。该结果表明1-gce复合材料对亚硝酸根和维生素c分别具有良好的还原性和氧化性。
[0060]
图9为本发明化合物1所制备电极材料在电催化前后的实验粉末x射线衍射图谱。催化反应后实验粉末x射线衍射图谱(1)与反应实验粉末x射线衍射图谱(2)基本一致,并与模拟图谱(3)很好的对应,表明催化剂具有良好的结构稳定性。
[0061]
对比例1
[0062]
制备方法同实施例1,区别仅在于:所选多酸基前驱体为k
14
[p2w
19o67
(h2o)]
·
24h2o;所得化合物的结构示意图如图10所示,由图10可知,所得化合物为零维结构(参考y.huo,m.l.tong,inorg.chem.2018,57,6773-6777)。
[0063]
对比例2
[0064]
制备方法同实施例1,区别在于:所选多酸基前驱体为k
14
[p2w
19o67
(h2o)]
·
24h2o、ph为7.4;所得化合物的结构示意图如图11所示,结构为三个三核稀土通过有机配体桥连的多核零维稀土结构(参考y.huo,m.l.tong,inorg.chem.2018,57,6773-6777)。
[0065]
对比例3
[0066]
制备方法同实施例1,区别在于:未引入有机配体、ph为4.4-5.0;所得化合物的结构示意图如图12所示,为多酸基单核稀土化合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1