一种Δ9(11)-坎利酮的制备方法与流程
一种
δ
9(11)-坎利酮的制备方法
技术领域
1.本发明涉及药物中间体制备方法,具体涉及一种δ9(11)-坎利酮的制备方法。
背景技术:
2.δ9(11)-坎利酮是依普利酮的中间体。2015年商艳梅等人在药学研究期刊中发表了一种以δ9(11)坎利酮为起始原料制备依普利酮的合成路线。上述方法减少了依普利酮合成过程中的副产物,提高了产率,也使得δ9(11)-坎利酮成为制备依普利酮一种重要的中间体。
3.对于δ9(11)-坎利酮的合成,现有技术有一步合成法和两步合成法。其中,一步合成法为2001年在杂志journal of fluorine chemistry中公开的基于1,1,2,2-四氟乙基-n,n-二甲胺以及基于yarovenko-raksha与ishikawa试剂作为氟化与脱水剂制备δ9(11)-坎利酮,其缺陷是这类氟化试剂生产成本较高,不利于工业化。此外,cn1633445a报道了一种使用pcl5在-50℃条件下一步合成δ9(11)-坎利酮的方法,但是该反应的反应温度需要-50℃,要求苛刻,也不利于工业化生产。目前,两步合成法文献报道多是甲磺酰基保护羟基,然后再以酸做溶剂消除合成δ9(11)坎利酮,其缺点是操作繁琐,且蒸馏除酸需要专用的耐腐蚀设备。
技术实现要素:
4.为了解决上述现有技术中存在的问题,本发明提供一种新的δ9(11)-坎利酮的制备方法。
5.具体地,本发明通过三氟甲磺酸酐做脱水剂,与11-α羟基坎利酮(如结构式1所示)反应,通过11-α羟基坎利酮与三氟甲磺酸酐发生羟基消除反应,一步合成δ9(11)坎利酮,其反应式如下:
[0006][0007]
本发明中,11-α羟基坎利酮与三氟甲磺酸酐的羟基消除反应在含有吡啶的溶剂中反应。优选地,所述羟基消除反应在含有吡啶的二氯甲烷溶剂中反应。其中,二氯甲烷与吡啶的体积比为10:(1~3),优选地为体积比约为10:3。
[0008]
本发明中,所述“约”是指以目标数值或目标比例为中心,允许不超过
±
10%的数值浮动范围,优选地,“约”是指以目标数值或目标比例为中心,允许不超过
±
5%的数值浮动范围。
[0009]
本发明中,首先将11-α羟基坎利酮加入溶剂中,得到含有11-α羟基坎利酮的溶液,
所述溶液中11-α羟基坎利酮的浓度为0.2~0.3mol/l,之后再将含有11-α羟基坎利酮的溶液的冷却,向溶液中加入三氟甲磺酸反应。
[0010]
优选地,本发明中,首先向含有吡啶的二氯甲烷溶剂中加入11-α羟基坎利酮,得到含有11-α羟基坎利酮的溶液,之后再向溶液中加入三氟甲磺酸反应。基于含有吡啶的二氯甲烷溶剂,加入11-α羟基坎利酮后其在溶液中的浓度为0.2~0.3mol/l。
[0011]
本发明中,以11-α羟基坎利酮的摩尔量基准(1eq.),11-α羟基坎利酮与三氟甲磺酸酐的投料当量比为1:(2~3)。
[0012]
本发明中,向含有11-α羟基坎利酮的溶液中加入三氟甲磺酸,进行羟基消除反应。该羟基消除反应的反应温度为-20℃~25℃,优选地,反应温度为-15℃~5℃,更优选地,反应温度为-15℃~-5℃,最优选地,反应温度为约-15℃。
[0013]
本发明的具体实施方式中,以11-α羟基坎利酮为原料与三氟甲磺酸酐发生羟基消除反应制备δ9(11)坎利酮的具体反应步骤如下:
[0014]
(1)将11-α羟基坎利酮加入到二氯甲烷和吡啶的混合液中得到11-α羟基坎利酮溶液;
[0015]
(2)将11-α羟基坎利酮溶液冷却,加入三氟甲磺酸酐,进行羟基消除反应;
[0016]
(3)反应结束后,经碱洗、去除溶剂得到δ9(11)坎利酮。
[0017]
其中,步骤(2)中先将11-α羟基坎利酮溶液冷却至-20℃~25℃,加入三氟甲磺酸酐,再保温反应。所述保温反应是在保持11-α羟基坎利酮溶液冷却后的温度的条件下,加入三氟甲磺酸酐进行反应。优选地,11-α羟基坎利酮溶液的冷却温度为-15℃~5℃,更优选地,冷却温度为-15℃~-5℃,最优选地,冷却温度为约-15℃。
[0018]
其中,步骤(3)中,所述碱洗为通过饱和的碳酸氢钠水溶液对步骤(2)的产物洗涤;碱洗后取有机层,并进一步去除有机层中溶剂,得到δ9(11)坎利酮。所述去除溶剂的方法为取碱洗后的有机相通过减压蒸馏去除溶剂,所述减压蒸馏的温度为35℃~50℃,压力为0.01~0.1atm,优选地,减压蒸馏的温度为约45℃。
[0019]
本发明中,还包括对采用本发明的方法制备δ9(11)坎利酮进一步提纯、包装的步骤。所述提纯包括但不限于重结晶、大孔树脂过柱分离提纯等。所述包装,可举例地是将通过本发明的方法合成得到的δ9(11)坎利酮或经进一步提纯的δ9(11)坎利酮出于方便存储、运输、给药、计量的目的而对δ9(11)坎利酮进行的包装。上述提纯和/或包装得到产品均应理解为由本专利保护的方法直接获得的产品。
[0020]
在本发明通过实验发现三氟甲磺酸酐做脱水剂,吡啶做碱,与11-α羟基坎利酮反应,羟基消除脱水一步合成δ9(11)坎利酮,-15℃~25℃即可反应。反应条件温和,后处理简单,对设备要求不高。与现有技术相比,本发明直接一步完成。且本发明相对于现有技术中使用五氯化磷的反应,反应条件更温和。本发明使用三氟甲磺酸酐做试剂,比ishikawa试剂等氟化试剂更廉价易得,具有工业化成本低等优点。
附图说明
[0021]
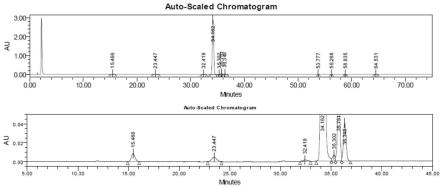
图1:实施例1产物的hplc谱图。
[0022]
图2:实施例2产物的hplc谱图。
[0023]
图3:实施例3产物的hplc谱图。
[0024]
图4:实施例4产物的hplc谱图。
[0025]
图5:实施例5产物的hplc谱图。
具体实施方式
[0026]
为使本发明的目的、技术方案及优点更加清楚、明确,以下参照附图并举实施例对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0027]
实施例1:δ9(11)坎利酮的合成
[0028]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(90ml)的溶液中,降温至-15℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕保温反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体25.4g。纯度95.47%(hplc),收率89.2%,产物hplc谱图如图1所示,hplc数据如下:
[0029]
表1:实施例1产物hplc数据
[0030][0031]
实施例2:δ9(11)坎利酮的合成
[0032]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(90ml)的溶液中,降温至-5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕保温反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体24.9g。纯度95.64%(hplc),收率87.4%,产物hplc谱图如图2所示,hplc数据如下:
[0033]
表2:实施例2产物hplc数据
[0034][0035]
实施例3:δ9(11)坎利酮的合成
[0036]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(90ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕保温反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体25.1g。纯度94.95%(hplc),收率87.0%,产物hplc谱图如图3所示,hplc数据如下:
[0037]
表3:实施例3产物hplc数据
[0038][0039]
实施例4:δ9(11)坎利酮的合成
[0040]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(90ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕升温至15℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体21.5g。纯度92.64%(hplc),收率75.5%,产物hplc谱图如图4所示,hplc数据如下:
[0041]
表4:实施例4产物hplc数据
[0042][0043]
实施例5:δ9(11)坎利酮的合成
[0044]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(90ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕升温至25℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体18.3g。纯度90.52%(hplc),收率64.2%,产物hplc谱图如图5所示,hplc数据如下:
[0045]
表5:实施例5产物hplc数据
[0046][0047]
在实施例1~5中可以看出,反应温度从-15℃到5℃之间产物收率在85%~90%之间,-15℃时hplc 95.47%为最佳值。当反应温度维持在15℃和25℃反应时,杂质明显上升,收率降低,产物hplc降到90.52%。
[0048]
对比例1:用乙酸酐替换三氟甲磺酸酐
[0049][0050]
将11-α羟基坎利酮(10g,29.5mmol)加入到二氯甲烷(100ml)和吡啶(30ml)的溶液中,降温至5℃,滴加乙酸酐(6.1g,59.1mmol)。滴加完毕升温至25℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。
过滤得淡黄色固体11-乙酰氧基坎利酮9.3g,无法一步合成得到δ9(11)坎利酮。hplc 97.56%,收率79.1%。
[0051]
对比例2:用三氟乙酸酐替换三氟甲磺酸酐
[0052][0053]
将11-α羟基坎利酮(10g,29.5mmol)加入到二氯甲烷(100ml)和吡啶(30ml)的溶液中,降温至5℃,滴加三氟乙酸酐(6.7g,59.1mmol)。滴加完毕升温至25℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体11-三氟乙酰氧基坎利酮10.6g,无法一步合成得到δ9(11)坎利酮。hplc 98.63%,收率83.5%。
[0054]
对比例3:用苯磺酰氯替换三氟甲磺酸酐
[0055][0056]
将11-α羟基坎利酮(10g,29.5mmol)加入到二氯甲烷(100ml)和吡啶(30ml)的溶液中,降温至5℃,滴加苯磺酰氯(10.4g,59.1mmol)。滴加完毕升温至25℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体11-苯磺酰氧基坎利酮10.9g,无法一步合成得到δ9(11)坎利酮。hplc 95.46%,收率78.2%。
[0057]
对比例4:将反应的溶剂体系换为:二氯甲烷与三乙胺
[0058]
将11-α羟基坎利酮(10g,29.5mmol)加入到二氯甲烷(100ml)和三乙胺(30ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(16.7g,59.1mmol)。滴加完毕升温至25℃左右反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。正己烷:乙酸乙酯=5~3:1硅胶粉200~300目过柱纯得到δ9(11)坎利酮3.2g,收率33.7%,hplc 98.26%。
[0059]
对比例5:将反应的溶剂体系换为:二氯甲烷与n,n-二乙基异丙胺
[0060]
将11-α羟基坎利酮10g,按照对比实施例4中操作,将三乙胺替换为n,n-二乙基异丙胺(30ml),最终过柱得到δ9(11)坎利酮,3.6g,收率37.9%。
[0061]
对比例6:将反应的溶剂体系换为:二氯甲烷与n-甲基吗啉
[0062]
将11-α羟基坎利酮10g,按照对比实施例4中操作,将三乙胺替换为n-甲基吗啉(30ml),过柱得到δ9(11)坎利酮,2.4g,收率25.3%。
[0063]
对比例7:将反应的溶剂体系换为:二氯甲烷与咪唑
[0064]
将11-α羟基坎利酮10g,按照对比实施例4中操作,将三乙胺替换为咪唑(30g),过
柱得到δ9(11)坎利酮,1.2g,收率12.6%。
[0065]
对比例8:将反应的溶剂体系换为:甲苯和吡啶合成δ9(11)坎利酮
[0066]
将11-α羟基坎利酮10g,按照对比实施例4中操作,将二氯甲烷替换为甲苯(100ml),过柱得到δ9(11)坎利酮,4.5g,收率47.4%。
[0067]
对比例9:二氯甲烷和吡啶10:1合成δ9(11)坎利酮
[0068]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(30ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕保温反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体17.6g。收率61.8%,hplc95.63%。
[0069]
对比例10:二氯甲烷和吡啶10:0.7合成δ9(11)坎利酮
[0070]
将11-α羟基坎利酮(30g,84.1mmol)加入到二氯甲烷(300ml)和吡啶(21ml)的溶液中,降温至5℃,滴加三氟甲磺酸酐(47.5g,168.2mmol)。滴加完毕保温反应16h,倒入冰水中,分液。有机层用饱和碳酸氢钠洗涤至碱性,分液,有机层45℃减压浓缩至固体大量析出。过滤得淡黄色固体12.6g。收率44.2%,hplc93.52%。
[0071]
通过对比例9和10吡啶降到<10:1时,反应开始变杂,产物收率开始降低,hplc也降到93.52%。
[0072]
本发明中,所述hplc是代表高效液相色谱(high performance liquid chromatography\hplc)。本发明中所述纯度是基于hplc测得,也简要标记为hplc后加百分数用于表达纯度。以对比例1为例,hplc 97.56%即表达通过hplc测定的目标产物纯度为97.56%。本发明中,所述收率是基于反应产物与理论产物的摩尔比。以下表6、表7为本发明中使用的液相色谱的条件。
[0073]
表6 hplc液相色谱条件1
[0074][0075][0076]
表7 hplc液相色谱条件2
[0077][0078]
本发明通过实验筛选,乙酸酐,三氟乙酸酐和苯磺酰氯,都是生成相应的11位酯,无法一步脱水消除。而三氟甲磺酸酐做脱水剂,吡啶做碱,与11-α羟基坎利酮反应,羟基消除脱水一步合成δ9(11)坎利酮,-15℃~25℃即可反应。反应条件温和,后处理简单,对设备要求不高。其他碱类如三乙胺,n,n-二乙基异丙胺,n-甲基吗啉和咪唑做碱,均有大量杂质生成,收率较低。
[0079]
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1