一种新型呸啶型配合物的合成方法及应用
1.本发明属于能源与均相催化技术领域,尤其涉及一种新型呸啶型配合物的合成方法及在催化二氧化碳加氢制甲酸盐中的应用。
背景技术:
2.现今,由煤、石油、天然气等含碳化石燃料大量燃烧所产生的co2排放量仍处于上升趋势。这带来了全球气候变暖、土地沙漠化等严峻环境问题。因此,通过化学反应将co2转化为清洁燃料和高附加值化学产品已经成为全球共同的目标。
3.另外,寻求新的可再生能源也至关重要。氢能是一种清洁的可再生能源,其燃烧产物只有水。且相同条件下,氢气的质量能量密度是汽油的3倍(33.3kw
·
h kg-1
),但标况下,氢气(2.5w
·
h l-1
)的体积能量密度仅是汽油(8.07kw
·
h l-1
)的三千分之一。(d.mellmann,p.sponholz,h.junge,m.beller,chem.soc.rev.,2016,45,3954—3988.)因此,对氢气进行大量、高效储存是实现氢经济的先决条件。
4.甲酸不仅是重要的有机合成中间体,而且是良好的液态储氢材料。具有储氢密度高(常温常压下53g h
2 l-1
)、运输方便、热力学稳定等优点。(s.chatterjee,i.dutta,y.w.lum,z.p.lai,k.-w.huang,energy environ.sci.,2021,14,1194
–
1246.)研究co2加氢制甲酸有助于解决目前的能源及环境问题,实现经济上可行的co
2-hcooh的氢能循环。
5.近年来,对于过渡金属配合物催化二氧化碳加氢反应的研究逐渐增多。但是,许多配合物都使用了易被氧化、对环境有污染的磷配体,并且许多反应在高温高压下进行,条件苛刻。因此,开发稳定、高效、对环境污染小的催化剂很有必要。
6.本发明研究了一类新型呸啶型配合物,合成方法简单,结构新颖。通过实验探究发现,本发明所述配合物是一类能够在水及甲醇体系中催化co2加氢制甲酸盐的高效催化剂。
技术实现要素:
7.本发明所要解决的技术问题在于针对上述现有技术中温度过高、压力较大、活性较低、环境污染等不足,提供了一种呸啶型配合物催化二氧化碳加氢制甲酸盐的方法。可实现水及甲醇混合溶液中较低温度、低压下高效催化二氧化碳加氢制甲酸盐的反应。
8.一方面,本发明所述的合成方法包括如下步骤:
9.(1)在氮气保护下,将1,8-二氨基萘、2-吡啶甲酸衍生物或4-吡啶甲酸衍生物或咪唑-2-甲酸或1h-咪唑-4-甲酸或2-甲酸吡嗪或2-嘧啶甲酸、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edci)、1-羟基苯并三唑(hobt)按照摩尔比1:1:1.2:1.2置于二氯甲烷中,原料总浓度为0.22-0.44mol/l,室温下搅拌24h。经水洗、干燥、过滤、浓缩及柱层析纯化或薄层色谱法纯化后得到对应的配体;
10.(2)氮气保护条件下,将上述配体与金属前体[cp*ircl2]2或[ru(η-c6h6)cl2]2或[cp*ir(h2o)3]so4或[ru(η-c6h6)(h2o)3]so4按照摩尔比1:1加入到甲醇或水中,配体与金属前体的总浓度为0.003-0.012mol/l,在60℃或25℃搅拌反应12h;经过滤、浓缩、重结晶纯化
后得到目标配合物,其结构式如下式(i)、(ii):
[0011][0012]
式中:r1、r2相互独立,相同或不同,为h、oh、ch3、或och3;
[0013]
x1、x2相互独立,相同或不同,为c或n;
[0014]
l=cl或h2o;
[0015]
ar=五甲基环戊二烯基(cp*)、苯、六甲基苯或对甲基异丙基苯;
[0016]
m=ir或ru;
[0017]
n=1或2;
[0018]
w=氯离子、硫酸根离子或四氟化硼阴离子。
[0019]
将本发明的配合物应用于催化二氧化碳加氢制甲酸盐。步骤如下:
[0020]
(1)配制0.5-2.0mol/l的khco3或nahco3或k2co3或cs2co3或na2co3溶液,溶剂为水与甲醇的混合溶液或超纯水;将配好的溶液盛于单口瓶,储存备用;
[0021]
(2)将新型呸啶型配合物溶解于甲醇中,新型呸啶型配合物的浓度为5μmol/ml;
[0022]
(3)按照体积比为50-2000:1向30ml反应釜中加入碱溶液及5μmol/ml配合物溶液;密封反应釜;使用体积比为1:1的h2和co2的混合气置换3次,加压至1-3mpa,升温80-120℃,搅拌反应1-48h;
[0023]
(4)反应结束后,将反应液使用滤膜过滤,使用高效液相色谱,利用标准曲线,对于得到的峰面积进行甲酸盐浓度进行计算,进而计算反应的ton值。
[0024]
本发明的有益效果:
[0025]
1、本发明制得的金属配合物催化剂结构新颖,是首例用于二氧化碳加氢制取甲酸盐领域的呸啶结构催化剂,制备方法比较简单,稳定性好,且一定程度上改善了现有技术中反应条件温度过高、压力较大、环境污染等不足。
[0026]
2、本发明中所述的配合物催化二氧化碳加氢制甲酸盐方法易于操作,且以co2作为碳源能够使碳排放量有所减少,符合绿色化学理念,有利于环境保护。
附图说明
[0027]
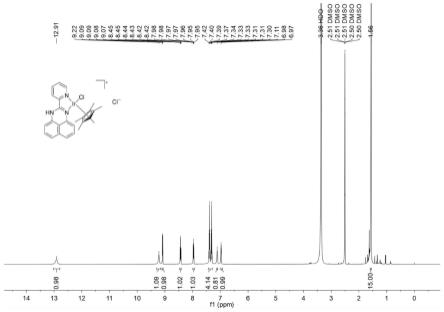
图1是式(i)中的r1=h,r2=h,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子的配合物1的1h nmr谱图。
[0028]
图2是配合物1的
13
c nmr谱图。
[0029]
图3是式(i)中的r1=h,r2=h,m=ru,ar=苯,l=cl,n=1,w=氯离子的配合物2的1h nmr谱图。
[0030]
图4是配合物2的
13
c nmr谱图。
[0031]
图5是式(i)中的r1=h,r2=h,m=ru,ar=对甲基异丙基苯,l=cl,n=1,w=氯离子的配合物3的1h nmr谱图。
[0032]
图6是式(i)中的r1=oh,r2=h,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子的配合物4的1h nmr谱图。
[0033]
图7是式(ii)中的x1=c,x2=n,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子的配合物6的1h nmr谱图。
[0034]
图8是配合物6的
13
c nmr谱图。
[0035]
图9是配合物4催化二氧化碳加氢制甲酸盐在压力为1mpa(体积比h2/co2=1:1),催化剂用量0.5μmol,10ml的1mol/l nahco3溶液(溶剂中水:甲醇体积比为6:4),24h的条件下,ton随温度变化关系图。
[0036]
图10是配合物4催化二氧化碳加氢制甲酸盐在120℃,1mpa(体积比h2/co2=1:1),10ml的1mol/l nahco3溶液(溶剂中水:甲醇体积比为6:4),24h的条件下,ton值随配合物4物质的量变化关系曲线。
[0037]
图11是配合物4催化二氧化碳加氢制甲酸盐在120℃,催化剂用量0.1μmol,10ml的1mol/l nahco3溶液(溶剂中水:甲醇体积比为6:4),24h的条件下,ton值随反应压力变化关系曲线。
具体实施方式
[0038]
为使技术方案和优点更加清楚,以下将对技术方案的具体实施方式进行进一步说明。
[0039]
实施例1
[0040]
式(i)中r1=h,r2=h,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子,标记为配合物1。其合成的反应方程式如下:
[0041][0042]
配合物1的制备方法如下:
[0043]
称取1,8-二氨基萘(1.0mmol)、2-吡啶甲酸(1.0mmol)、edci(1.2mmol)、hobt(1.2mmol)加入到50ml双口瓶中,一端密封,一端连接冷凝管后与双排管相连,置换氮气,并在氮气的氛围下加入10ml二氯甲烷,室温搅拌反应24h。反应停止后,使用20ml去离子水洗
涤三次,除去水溶性杂质,收集下层有机相,加入无水硫酸钠干燥,然后过滤,旋转蒸发除去溶剂,经过真空干燥得到粗产物。使用柱层析法纯化后,对所得溶液旋转蒸发除去溶剂,再对所得固体真空干燥,得到亮红色固体配体1。收率为75%。
[0044]
称取配体1(0.08mmol)、[cp*ircl2]2(0.04mmol),加入50ml两口瓶中,一瓶口连接球形冷凝管。置换氮气后,在通氮气的条件下,向瓶中加入20ml甲醇溶液。升温至65℃,反应12小时,可见反应液由鲜红色变为紫红色。将反应后的液体旋蒸出甲醇,向固体中加入大量异丙醇后,用膜过滤装置过滤反应液,除去不溶性杂质。将所得液体浓缩,再加入正己烷,析出紫色固体,膜过滤,收集固体。向瓶中加入异丙醇,再浓缩,用正己烷重结晶,收集析出的固体。重复上述步骤直至没有固体析出。得到紫红色固体配合物1,产率为93%。
[0045]
配合物1的核磁共振氢谱如图1所示,核磁共振碳谱如图2所示,具体谱图数据如下:1h nmr(500mhz,dmso-d6)δ12.33(s,1h),8.40(d,j=7.2hz,1h),8.16(t,j=8.0hz,1h),7.38(d,j=6.3hz,3h),7.32
–
7.28(m,2h),7.04(dd,j=6.5,2.0hz,1h),6.98(s,1h),1.54(s,15h).
13
c nmr(126mhz,dmso-d6)δ153.85,140.75,134.91,130.83,128.93,128.61,126.32,123.10,122.60,121.45,117.41,107.15,88.98,9.04.
[0046]
实施例2
[0047]
式(i)中的r1=h,r2=h,m=ru,ar=苯,l=cl,n=1,w=氯离子,标记为配合物2。其合成的反应方程式如下:
[0048][0049]
配合物2的制备方法如下:
[0050]
称取配体1(0.08mmol)、二氯苯基钌(ⅱ)二聚体(0.04mmol),加入50ml两口瓶中,一瓶口连接球形冷凝管。在瓶外包一层锡纸后,置换氮气,在通氮气的条件下,向瓶中加入20ml甲醇溶液。升温至65℃,反应12小时。可见反应液由鲜红色变为紫红色。将反应后的液体减压蒸馏除去甲醇,向固体中加入大量异丙醇后,用膜过滤装置过滤反应液,除去不溶性杂质。将所得液体浓缩,再加入正己烷,析出灰黑色固体,膜过滤,收集固体。向瓶中加入异丙醇,再浓缩,用正己烷重结晶,收集析出的固体。重复上述步骤直至没有固体析出。得到灰黑色固体配合物2,产率为76%。
[0051]
配合物2的核磁共振氢谱如图3所示,核磁共振碳谱如图4所示,具体谱图数据如下:1h nmr(500mhz,dmso-d6)δ12.53(s,1h),9.65(d,j=5.5hz,1h),8.98(s,1h),8.40(t,j=7.9hz,1h),7.93(t,j=6.6hz,1h),7.40(q,j=5.4hz,3h),7.36
–
7.27(m,2h),7.02(s,1h),6.14(s,6h).
13
c nmr(126mhz,dmso-d6)δ157.03,143.04,140.27,135.15,134.93,129.38,128.70,122.74,122.25,121.10,117.74,106.44,87.08.
[0052]
实施例3
[0053]
式(i)中的r1=h,r2=h,m=ru,ar=对甲基异丙基苯,l=cl,n=1,w=氯离子,标记为配合物3。其合成的反应方程式如下:
[0054][0055]
配合物3的制备方法如下:
[0056]
称取配体1(0.08mmol)、二氯双(4-甲基异丙基苯基)钌(ⅱ)(0.04mmol),加入50ml两口瓶中,一瓶口连接球形冷凝管。在瓶外包一层锡纸后,置换氮气,在通氮气的条件下,向瓶中加入20ml甲醇溶液。升温至65℃,反应12小时。可见反应液由鲜红色变为紫红色。将反应后的液体减压蒸馏除去甲醇,向固体中加入大量异丙醇后,用膜过滤装置过滤反应液,除去不溶性杂质。将所得液体浓缩,再加入正己烷,析出灰黑色固体,膜过滤,收集固体。向瓶中加入异丙醇,再浓缩,用正己烷重结晶,收集析出的固体。重复上述步骤直至没有固体析出。得到灰黑色固体配合物3,产率为83%。
[0057]
配合物3核磁共振氢谱如图5所示,具体谱图数据如下:1h nmr(500mhz,dmso-d6)δ12.54(s,1h),9.53(d,j=5.5hz,1h),8.97(s,1h),8.39(t,j=7.8hz,1h),7.92(t,j=6.6hz,1h),7.46
–
7.18(m,5h),6.99(s,1h),6.02(dd,j=13.0,6.1hz,2h),5.91(dd,j=19.9,6.1hz,2h),2.59(p,j=6.9hz,1h),2.28(s,3h),1.07(d,j=6.9hz,3h),1.00(d,j=6.9hz,3h).
[0058]
实施例4
[0059]
式(i)中的r1=oh,r2=h,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子,标记为配合物4。其合成的反应方程式如下:
[0060][0061]
配合物4的制备方法如下:
[0062]
称取6-羟基吡啶-2-甲酸(0.5mmol)、1,8-二氨基萘(0.5mmol)、edci(0.6mmol)、hobt(0.6mmol)加入到50ml双口瓶中,一端密封,一端连接冷凝管后与双排管相连,置换氮气,并在氮气的氛围下加入10ml二氯甲烷,室温搅拌反应24h。在反应过程中,溶液变浑浊,
出现絮状物,且由鲜红色变为红褐色。反应停止后,使用50ml去离子水洗涤五次,除去水溶性杂质及红褐色絮状物,收集下层有机相,上层水相使用5ml二氯甲烷反复萃取,至萃取有机相薄层色谱层析无明显紫外吸收显色,合并有机相,加入无水硫酸钠干燥。然后过滤,旋转蒸发除去溶剂,经过真空干燥得到暗红色固体配体2。收率为73%。
[0063]
称取配体2(0.04mmol)、[cp*ircl2]2(0.02mmol),加入50ml两口瓶中,一瓶口连接球形冷凝管。置换氮气后,在通氮气的条件下,向瓶中加入15ml甲醇溶液,65℃反应12小时。反应停止后,旋转蒸发除去溶剂。向固体中加入大量异丙醇,超声,膜过滤除去固体。浓缩异丙醇,加入正己烷,有暗红色固体析出,膜过滤收集固体。再将瓶壁上的固体用异丙醇溶解,浓缩,再加入正己烷,过滤收集固体,重复上述步骤,得到紫红色固体配合物4。产率82%。
[0064]
对配合物4的结构进行了表征,核磁共振氢谱如图6所示,具体谱图数据如下为:1h nmr(500mhz,dmso-d6)δ12.91(s,1h),9.22(s,1h),9.08(dd,j=5.7,1.4hz,1h),8.43(td,j=7.9,1.4hz,1h),7.97(ddd,j=7.2,5.5,1.3hz,1h),7.44
–
7.28(m,4h),7.11(s,1h),1.56(s,15h).
[0065]
实施例5
[0066]
式(i)中的r1=h,r2=och3,m=ir,ar=五甲基环戊二烯基(cp*),l=h2o,n=2,w=硫酸根离子,标记为配合物5。其合成的反应方程式如下:
[0067][0068]
配合物5的制备方法如下:
[0069]
称取4-甲氧基吡啶-2-甲酸(0.5mmol)、1,8-二氨基萘(0.5mmol)、edci(0.6mmol)、hobt(0.6mmol)加入到50ml双口瓶中,一端密封,一端连接冷凝管后与双排管相连,置换氮气,并在氮气的氛围下加入10ml二氯甲烷,室温搅拌反应24h。反应停止后,使用50ml去离子水洗涤五次,除去水溶性杂质,收集下层有机相,旋转蒸发除去溶剂后,得到粗产物。再通过柱层析色谱分离纯化(展开剂二氯甲烷:甲醇=20:1)得到红色固体配体3。收率为87%。
[0070]
称取配体3(0.1mmol)、[cp*ir(oh2)3](so4)(0.1mmol)加入25ml两口瓶中,置换氮气后,在通氮气的条件下,向瓶中加入15ml超纯水,室温搅拌反应12h。反应停止后,水膜过滤除去少量不溶物,收集滤液,旋转蒸发除去溶剂。再用甲醇/乙醚重结晶三次,得到紫红色配合物5。产率为75%。
[0071]
实施例6
[0072]
式(ii)中的x1=c,x2=n,m=ir,ar=五甲基环戊二烯基(cp*),l=cl,n=1,w=氯离子,标记为配合物6。其合成的反应方程式如下:
[0073][0074]
配合物6的制备方法如下:
[0075]
称取2-嘧啶甲酸(0.5mmol)、1,8-二氨基萘(0.5mmol)、edci(0.6mmol)、hobt(0.6mmol)加入到50ml双口瓶中,一端密封,一端连接冷凝管后与双排管相连,置换氮气,并在氮气的氛围下加入10ml二氯甲烷,室温搅拌反应24h。反应停止后,溶液呈现暗红色。反应停止后,使用20ml去离子水洗涤三次,除去水溶性杂质,收集下层酒红色有机相,加入无水硫酸钠干燥,然后过滤,旋转蒸发除去溶剂,经过真空干燥得到粗产物。使用薄层色谱法纯化后,刮下硅胶用二氯甲烷浸泡,过滤后对溶液先旋转蒸发除去溶剂,再对所得固体使用正己烷重结晶。最后将固体真空干燥,得到暗红色固体配体4。收率为68%。
[0076]
称取配体4(0.04mmol)、[cp*ircl2]2(0.02mmol),加入50ml两口瓶中,置换氮气后,在通氮气的条件下,向瓶中加入15ml甲醇溶液,65℃反应12小时。反应停止后,旋转蒸发除去溶剂。向固体中加入大量异丙醇,超声,膜过滤除去固体。浓缩异丙醇,加入正己烷,有紫红色固体析出,膜过滤收集固体。再将瓶壁上的固体用异丙醇溶解,浓缩,再加入正己烷,过滤收集固体,重复上述步骤,得到紫红色固体配合物6。产率91%。
[0077]
配合物6的核磁共振氢谱如图7所示,核磁共振碳谱如图8所示,具体谱图数据为:1h nmr(500mhz,dmso-d6)δ11.01(s,1h),9.03(d,j=4.9hz,2h),7.72(t,j=4.8hz,1h),7.16(t,j=7.7hz,2h),7.06(d,j=8.4hz,2h),6.73(d,j=7.3hz,2h).
13
c nmr(126mhz,dmso-d6)δ158.20,157.94,150.51,135.61,129.01,123.20,122.85.
[0078]
应用例1
[0079]
以本发明举例制备的配合物1为例,说明二氧化碳加氢制甲酸盐反应的过程:
[0080]
(1)配制2mol/l的khco3的超纯水溶液,将配好的溶液盛于单口瓶,储存备用;
[0081]
(2)称取5μmol配合物1,再向其中加入1ml甲醇进行溶解;
[0082]
(3)向反应釜中加入磁子、10ml上述khco3溶液以及200μl上述5μmol/ml配合物1的甲醇溶液,密封反应釜。使用混合气(h2/co2=1:1)置换3次,加压至1mpa,升温至80℃,搅拌反应24h;
[0083]
(4)反应结束后,将反应液用滤膜过滤。使用高效液相色谱,利用标准曲线,对于得
到的峰面积进行甲酸盐浓度进行计算,进而计算反应的ton值为246。
[0084]
应用例2-6
[0085]
应用例2-6与应用例1的不同之处在于,用于催化二氧化碳加氢制甲酸盐的配合物不一致,详见表1。
[0086]
表1应用例2-6试验数据表
[0087][0088][0089]
应用例7
[0090]
应用例7与应用例1的不同之处在于,将应用例1中的配合物1换成了配合物4;将应用例1中2mol/l khco3的超纯水溶液换成相同浓度的nahco3、k2co3、cs2co3、na2co3超纯水溶液;将应用例1中加入反应釜的催化剂用量换为100μl。
[0091]
应用例8
[0092]
应用例8与应用例1的不同之处在于,将应用例1中的配合物1换成了配合物4;将应用例1中2mol/l khco3的超纯水溶液换成1mol/l nahco3溶液,且溶剂中水:甲醇体积=6:4;将应用例1中加入反应釜中的催化剂用量换为100μl。改变温度,进行实验。
[0093]
应用例9
[0094]
应用例9与应用例1的不同之处在于,将应用例1中的配合物1换成了配合物4;将应用例1中2mol/l khco3的超纯水溶液换成1mol/l nahco3溶液,且溶剂中水:甲醇体积=6:4;将应用例1中温度换为120℃。改变配合物4的量,进行实验。
[0095]
应用例10
[0096]
应用例10与应用例1的不同之处在于,将应用例1中的配合物1换成了配合物4;将应用例1中2mol/l khco3的超纯水溶液换成1mol/l nahco3溶液,且溶剂中水:甲醇体积=6:4;将应用例1中加入反应釜中的催化剂用量换为20μl。改变压力,进行实验。
[0097]
表2应用例7-10反应参数及试验结果汇总表
[0098][0099]
结合应用实例7及表2可以看出,在较弱的碱中,配合物的催化活性就能够达到较高水平。结合实例8及表2可以看出,随着温度的升高,ton值不断增大,所制备的甲酸盐越多(图9)。结合实例9及表2可以看出,ton随着催化剂的用量减少呈现出先升高后降低的趋势(图10),在较低的催化剂用量下,即0.1μmol下,ton达到较高值。结合实例10及表2可以看出,压力越大,能够制得的甲酸盐产量越高。(图11)
[0100]
综上所述,本技术制备方法所制得的新型呸啶型配合物结构新颖,ton值较高,应用于催化加氢制甲酸盐中,制得的甲酸盐产量高,对于二氧化碳的资源化利用提供了一种新的思路。
[0101]
需要说明的是,在本发明的举例中,采用的惰性气体均为氮气,以便更好说明本发明的技术方案,本领域技术人员还可以选用其他惰性气体作为保护气体。本发明提供的具体实施方式只是本发明的实例,并不构成本发明保护范围的限定。对于本领域的技术人员
而言,在不付出创造性劳动的前提下,依据本发明方案所扩展出的任何其他实施方式都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1