通过C-H键官能团化合成氟代氮杂芳烃化合物的方法
通过c
−
h键官能团化合成氟代氮杂芳烃化合物的方法
技术领域
1.本发明涉及催化合成技术领域。更具体地,涉及一种通过c
−
h键直接官能团化合成氟代氮杂芳烃化合物的方法。
背景技术:
2.氟代氮杂芳烃化合物是一类重要的有机氟化合物,广泛存在于医药、农药、功能材料及精细化学品等功能分子中,在药物化学、合成化学和材料科学等领域均有重要的应用(chem. rev. 2015,115, 612-633;j. med. chem. 2009,52, 7360-7363.)。因此,建立合成氟代氮杂芳烃化合物的方法具有重要的合成意义。目前,合成氟代氮杂芳烃化合物的技术主要包括balz-scheiemann反应(chem. ber. 1927,60, 1186-1190;j. heterocyclic chem. 2001,38, 793-808.)和基于芳香亲核取代反应(snar)的卤素氟-氯或硝基交换反应(acc. chem. res. 2020,53, 2372-2383;chem. soc. rev. 1999,28, 225-231;angew. chem. int. ed. 2006,45, 2720-2725.)。然而,balz-scheiemann反应通常需要强酸和强氧化剂,反应过程有易爆的重氮盐生成,存在安全隐患,且反应的重复性较差,同时,该反应生成了大量的废液或固体废弃物如nabf4、nacl或hcl;在芳香亲核取代反应中,反应底物需进行预活化,且要连接强吸电子性质的取代基,底物范围受限,为不影响氟化物的亲核性和避免副产物生成,反应需要在严格无水条件下进行。因此,发展条件温和、操作简单、安全、绿色、步骤经济和原子经济的合成氟代氮杂芳烃化合物的方法十分必要。
3.通过c
−
h键官能团化合成氟代氮杂芳烃化合物的方法是一类极具吸引力的合成方法。该方法减少了各种试剂和原料的预先官能团化,具有步骤少、原子经济、操作简单等特点。然而,直接实现氮杂芳烃的c(sp2)
−
h氟化反应存在很大挑战:(1)氮杂芳烃的c(sp2)
−
h键具有很强的惰性,且该类化合物与大多数的亲核性f-化合物的极性不匹配,难以直接发生亲核反应;(2)氮杂芳烃具有缺电子芳香性,其电性也与f
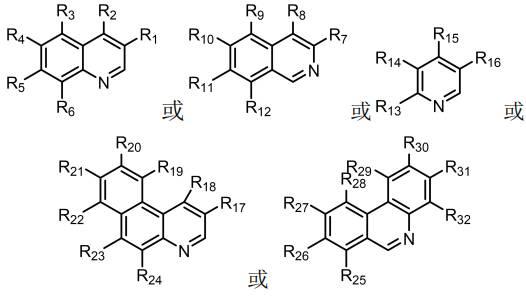
+
亲电氟试剂的极性不匹配,当用亲电氟试剂与它发生亲电进攻时,导致能量更高,反应也不易进行。
4.早期,为实现氮杂芳烃c(sp2)
−
h氟化反应,化学家们通常使用高活性的f2作为氟源,与氮杂芳香化合物反应(tetrahedron lett. 1987,28, 255-258;j. fluorine chem. 1999,100, 63-73)。2013年,来自加利福尼亚大学的john f. hartwig课题组在science上发表了一篇极具开创性的工作,他们利用agf2作催化剂,实现了吡啶c(sp2)
−
h氟化反应,为氟代氮杂芳烃化合物的合成提供了一种高效的方法(science 2013,342, 956-960.)。然而,现有的通过c
−
h键直接官能团化合成氟代氮杂芳烃化合物的技术需要使用贵金属催化剂和高反应性、高毒性和强腐蚀性的f2,这极大限制了其应用。
5.因此,需要发展一种无需金属催化剂的参与,操作简单、安全、绿色、步骤经济和原子经济的合成氟代氮杂芳烃化合物的方法。
技术实现要素:
6.本发明的目的在于提供一种通过c
−
h键官能团化合成氟代氮杂芳烃化合物的方
法。
7.为达到上述目的,本发明采用下述技术方案:一种通过c
−
h键官能团化合成氟代氮杂芳烃化合物的方法,该方法包括:在含有催化剂和溶剂的体系中,添加氮杂芳烃化合物、有机硅烷化合物和酸,在惰性气体氛围下,使用可见光照射进行反应,得到氟代氮杂芳烃化合物;所述催化剂为“n
−
f”试剂。本发明采用安全、稳定的“n
−
f”试剂作为催化剂和氟源,无需金属催化剂参与,底物无需预活化,具有反应条件温和、操作简单、安全、绿色、高步骤经济性等优点,符合绿色发展的理念。
8.优选地,所述“n
−
f”试剂的结构式为:。
9.本发明利用安全、稳定的“n
−
f”试剂既作为反应的催化剂,又作为反应的氟源,在没有金属催化剂的参与下,实现氟代氮杂芳烃化合物的合成。
10.优选地,所述溶剂为有机溶剂或有机溶剂与水的混合液。
11.优选地,所述有机溶剂为乙腈、丙酮、乙酸乙酯、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、1,2-二氯乙烷、二氯甲烷、甲醇、乙醇、异丙醇和四氢呋喃中的一种或多种。
12.优选地,所述有机硅烷化合物为三乙基硅烷、三甲基硅烷、六甲基二硅烷中的一种或多种。
13.优选地,所述的有机硅烷的浓度是0.0002
ꢀ‑
1 mol/l。
14.优选地,所述酸选自甲酸、乙酸、盐酸、硫酸、硝酸、磷酸、三氟乙酸或苯甲酸中的一种或多种。
15.优选地,所述的酸浓度是0.0002
ꢀ‑
1 mol/l。
16.优选地,所述氮杂芳烃化合物在溶剂中的浓度为1
×
10-5
mmol/l~饱和浓度。所述氮杂芳烃化合物在溶剂中达到饱和浓度后,可以继续增加氮杂芳烃化合物的含量,只是在理论上没有经济价值。
17.优选地,所述氮杂芳烃化合物的结构式为:
式中,r1、r2、r3、r4、r5和r6中的一个或多个基团为h、och
3、
ch3ch
2、
(ch3)2ch、(ch3)3c、f、cl、br、nh
2、
oh、cn、no2或ph,其余的基团为h;r7、r8、r9、r
10
、r
11
和r
12
中的一个或多个基团为h、och
3、
ch3ch
2、
(ch3)2ch、(ch3)3c、f、cl、br、nh
2、
oh、cn、no2或ph,其余的基团为h;r
13
、r
14
、r
15
和r
16
中的一个或多个基团为h、och
3、
ch3ch
2、
(ch3)2ch、(ch3)3c、f、cl、br、nh
2、
oh、cn、no2或ph,其余的基团为h;r
17
、r
18
、r
19
、r
20
、r
21
、r
22
、r
23
和r
24
中的一个或多个基团为h、och
3、
ch3ch
2、
(ch3)2ch、(ch3)3c、f、cl、br、nh
2、
oh、cn、no2或ph,其余的基团为h;r
25
、r
26
、r
27
、r
28
、r
29
、r
30
、r
31
和r
32
中的一个或多个基团为h、och
3、
ch3ch
2、
(ch3)2ch、(ch3)3c、f、cl、br、nh
2、
oh、cn、no2或ph,其余的基团为h;具体的,通过c
−
h键直接官能团化合成氟代氮杂芳烃化合物的反应方程式如式(1)所示:式中,氮杂芳烃化合物的结构如前面所定义。
18.优选地,所述惰性气体为氮气、氩气。
19.优选地,所述可见光波长范围为300~800nm。
20.优选地,所述可见光的光源是led、氙灯、汞灯、太阳光。
21.优选地,所述可见光的光照时间为1-24小时。
22.优选地,所述合成氟代氮杂芳烃化合物的反应在室温下进行。
23.本发明的有益效果如下:1)本发明在可见光照射下,通过c
−
h键官能团化实现氟代氮杂芳烃化合物的合成。
24.2)本发明使用安全、稳定的“n
−
f”试剂作为催化剂和氟源,无需金属催化剂参与,反应条件温和,操作简单,安全,绿色,底物无需预活化,步骤少,原子经济。
附图说明
25.下面结合附图对本发明的具体实施方式作进一步详细的说明。
26.图1示出本发明实施例1中产物2-氟-4-甲基喹啉的氢谱图。
27.图2示出本发明实施例1中产物2-氟-4-甲基喹啉的碳谱图。
28.图3示出本发明实施例1中产物2-氟-4-甲基喹啉的氟谱图。
具体实施方式
29.为了更清楚地说明本发明,下面结合优选实施例和附图对本发明做进一步的说明。附图中相似的部件以相同的附图标记进行表示。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
30.另外,如无特殊说明,本发明中所用原料均可通过市售商购获得,本发明所记载的任何范围包括端值以及端值之间的任何数值以及端值或者端值之间的任意数值所构成的任意子范围。
31.实施例1步骤1)在10 ml试管中加入0.3 mmoln-氟代苯磺酰亚胺(nfsi)和3ml乙酸乙酯,得到混合溶液;步骤2)将0.1 mmol 4-甲基喹啉,0.2 mmol三乙基硅烷,0.3 mmol三氟乙酸加入到上述混合溶液中,得到反应液;步骤3)在氮气保护下,采用led蓝光(= 405 nm)照射步骤2)中的反应液24h;步骤4)旋干法除去反应溶剂,再用硅胶过柱分离,得到产物2-氟-4-甲基喹啉。
32.其中产物的收率为60%。核磁氢谱、碳谱、氟谱鉴定产物为2-氟-4-甲基喹啉,氢谱图见图1,碳谱图见图2,氟谱图见图3。
33.实施例2同实施例1,区别在于步骤1)中加入3 ml乙腈代替乙酸乙酯作为反应溶剂,所得产物的收率是50%。
34.实施例3同实施例1,区别在于步骤1)中加入3 ml丙酮代替乙酸乙酯作为反应溶剂,所得产物的收率是57%。
35.实施例4同实施例1,区别在于步骤1)中加入3 mln,n-二甲基甲酰胺代替乙酸乙酯作为反应溶剂,所得产物的收率是20%。
36.实施例5同实施例1,区别在于步骤1)中加入3 ml二甲基亚砜代替乙酸乙酯作为反应溶剂,所得产物的收率是16%。
37.实施例6同实施例1,区别在于步骤1)中加入3 ml1,2-二氯乙烷代替乙酸乙酯作为反应溶剂,所得产物的收率是12%。
38.实施例7同实施例1,区别在于步骤1)中加入3 ml二氯甲烷代替乙酸乙酯作为反应溶剂,所
得产物的收率是10%。
39.实施例8同实施例1,区别在于步骤1)中加入3 ml四氢呋喃代替乙酸乙酯作为反应溶剂,所得产物的收率是27%。
40.实施例9同实施例1,区别在于步骤2)中加入0.3 mmol甲酸代替三氟乙酸(tfa),所得产物的收率是53%。
41.实施例10同实施例1,区别在于步骤2)中加入0.3 mmol乙酸代替三氟乙酸(tfa),所得产物的收率是51%。
42.实施例11同实施例1,区别在于步骤2)中加入0.3 mmol苯甲酸代替三氟乙酸(tfa),所得产物的收率是51%。
43.实施例12同实施例1,区别在于步骤2)中加入0.3 mmol盐酸代替三氟乙酸(tfa),所得产物的收率是16%。
44.实施例13同实施例1,区别在于步骤1)中加入0.1 mmol n-氟代苯磺酰亚胺(nfsi)代替0.3 mmol n-氟代苯磺酰亚胺(nfsi),所得产物的收率是31%。
45.实施例14同实施例1,区别在于步骤1)中加入0.15 mmol n-氟代苯磺酰亚胺(nfsi)代替0.3 mmol n-氟代苯磺酰亚胺(nfsi),所得产物的收率是47%。
46.实施例15同实施例1,区别在于步骤1)中加入0.2 mmol n-氟代苯磺酰亚胺(nfsi)代替0.3 mmol n-氟代苯磺酰亚胺(nfsi),所得产物的收率是50%。
47.实施例16同实施例1,区别在于步骤1)中加入0.25 mmol n-氟代苯磺酰亚胺(nfsi)代替0.3 mmol n-氟代苯磺酰亚胺(nfsi),所得产物的收率是58%。
48.实施例17同实施例1,区别在于步骤1)中加入0.35 mmol n-氟代苯磺酰亚胺(nfsi)代替0.3 mmol n-氟代苯磺酰亚胺(nfsi),所得产物的收率是60%。
49.实施例18同实施例1,区别在于步骤2)中加入0.1 mmol三乙基硅烷代替0.2 mmol 三乙基硅烷,所得产物的收率是54%。
50.实施例19同实施例1,区别在于步骤2)中加入0.15 mmol三乙基硅烷代替0.2 mmol 三乙基硅烷,所得产物的收率是57%。
51.实施例20同实施例1,区别在于步骤2)中加入0.25 mmol三乙基硅烷代替0.2 mmol 三乙基
硅烷,所得产物的收率是56%。
52.实施例21同实施例1,区别在于步骤2)中加入0.3 mmol三乙基硅烷代替0.2 mmol 三乙基硅烷,所得产物的收率是53%。
53.实施例22同实施例1,区别在于步骤3)中采用led蓝光( = 430 nm)代替led蓝光(= 405 nm)照射反应液,所得产物的收率是48%。
54.实施例23同实施例1,区别在于步骤3)中采用led蓝光(= 450 nm)代替led蓝光(= 405 nm)照射反应液,所得产物的收率是34%。
55.实施例24同实施例1,区别在于步骤1)中加入1-氯甲基-4-氟-1,4-重氮化二环2.2.2辛烷双(四氟硼酸)盐(selectfluor)代替n-氟代双苯磺酰胺(nfsi),所得产物的收率是48%。
56.实施例25同实施例1,区别在于步骤1)中加入1-氟-4-甲基-1,4-二氮杂双环[2.2.2]辛烷四氟硼酸盐(selectfluor ii)代替n-氟代双苯磺酰胺(nfsi),所得产物的收率是48%。
[0057]
实施例26同实施例1,区别在于步骤2)中以0.1 mmol 3-甲基喹啉代替4-甲基喹啉,所得产物的收率是76%。
[0058]
实施例27同实施例1,区别在于步骤2)中以0.1 mmol 5-甲基喹啉代替4-甲基喹啉,所得产物的收率是20%。
[0059]
实施例28同实施例1,区别在于步骤2)中以0.1 mmol 6-甲基喹啉代替4-甲基喹啉,所得产物的收率是53%。
[0060]
实施例29同实施例1,区别在于步骤2)中以0.1 mmol 7-甲基喹啉代替4-甲基喹啉,所得产物的收率是51%。
[0061]
实施例30同实施例1,区别在于步骤2)中以0.1 mmol喹啉代替4-甲基喹啉,所得产物的收率是55%。
[0062]
实施例31同实施例1,区别在于步骤2)中以0.1 mmol 6-甲氧基喹啉代替4-甲基喹啉,所得产物的收率是47%。
[0063]
实施例32同实施例1,区别在于步骤2)中以0.1 mmol 4-氰基喹啉代替4-甲基喹啉,所得产物的收率是43%。
[0064]
实施例33
同实施例1,区别在于步骤2)中以0.1 mmol 6-氟喹啉代替4-甲基喹啉,所得产物的收率是35%。
[0065]
实施例34同实施例1,区别在于步骤2)中以0.1 mmol 6-氯喹啉代替4-甲基喹啉,所得产物的收率是53%。
[0066]
实施例35同实施例1,区别在于步骤2)中以0.1 mmol 5-氯喹啉代替4-甲基喹啉,所得产物的收率是50%。
[0067]
实施例36同实施例1,区别在于步骤2)中以0.1 mmol 4-氯喹啉代替4-甲基喹啉,所得产物的收率是47%。
[0068]
实施例37同实施例1,区别在于步骤2)中以0.1 mmol 6-溴喹啉代替4-甲基喹啉,所得产物的收率是48%。
[0069]
实施例38同实施例1,区别在于步骤2)中以0.1 mmol 3-溴喹啉代替4-甲基喹啉,所得产物的收率是63%。
[0070]
实施例39同实施例1,区别在于步骤2)中以0.1 mmol苯并喹啉代替4-甲基喹啉,所得产物的收率是51%。
[0071]
实施例40同实施例1,区别在于步骤2)中以0.1 mmol异喹啉代替4-甲基喹啉,所得产物的收率是43%。
[0072]
实施例41同实施例1,区别在于步骤2)中以0.1 mmol 6-甲基异喹啉代替4-甲基喹啉,所得产物的收率是51%。
[0073]
实施例42同实施例1,区别在于步骤2)中以0.1 mmol 4-甲氧基异喹啉代替4-甲基喹啉,所得产物的收率是47%。
[0074]
实施例43同实施例1,区别在于步骤2)中以0.1 mmol 5-甲氧基异喹啉代替4-甲基喹啉,所得产物的收率是43%。
[0075]
实施例44同实施例1,区别在于步骤2)中以0.1 mmol 4-溴异喹啉代替4-甲基喹啉,所得产物的收率是56%。
[0076]
实施例45同实施例1,区别在于步骤2)中以0.1 mmol 5-溴异喹啉代替4-甲基喹啉,所得产物的收率是40%。
[0077]
实施例46
同实施例1,区别在于步骤2)中以0.1 mmol 4-氯异喹啉代替4-甲基喹啉,所得产物的收率是43%。
[0078]
实施例47同实施例1,区别在于步骤2)中以0.1 mmol 6-氯异喹啉代替4-甲基喹啉,所得产物的收率是47%。
[0079]
实施例48同实施例1,区别在于步骤2)中以0.1 mmol 4-苯基异喹啉代替4-甲基喹啉,所得产物的收率是57%。
[0080]
实施例49同实施例1,区别在于步骤2)中以0.1 mmol 4-苯基吡啶代替4-甲基喹啉,所得产物的收率是50%。
[0081]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1