乌帕替尼中间体(3R,4S)-1-苄氧羰基-4-乙基吡咯烷-3-羧酸的合成方法与流程
乌帕替尼中间体(3r,4s)-1-苄氧羰基-4-乙基吡咯烷-3-羧酸的合成方法
技术领域
1.本发明涉及药物合成技术领域,更具体的是涉及乌帕替尼中间体合成技术领域。
背景技术:
2.乌帕替尼(upadacitinib)是一款由艾伯维开发的新型jak1抑制剂。2019年8月,乌帕替尼在美国获得全球首批,用于治疗对甲氨蝶呤(mtx)应答不足或不耐受的中度至重度活动性类风湿性关节炎(ra)成人患者。2019年12月,upadacitinib获得欧盟批准,用于治疗对一种或多种疾病修饰抗风湿药物(dmard)应答不足或不耐受的中度至重度ra成人患者。在ra中,乌帕替尼批准的剂量为15mg。
3.目前,艾伯维正在开发乌帕替尼治疗多种炎症性疾病,包括银屑病关节炎(psa)、ra、中轴型脊柱关节炎(axspa)、克罗恩病(cd)、特应性皮炎(ad)、溃疡性结肠炎(uc)、巨细胞动脉炎(gca)。业界对乌帕替尼的商业前景非常看好。医药市场调研机构evaluatepharma之前发布报告预测,rinvoq在2024年的全球销售额将达到25.7亿美元,成为全球第5大畅销抗风湿药物。
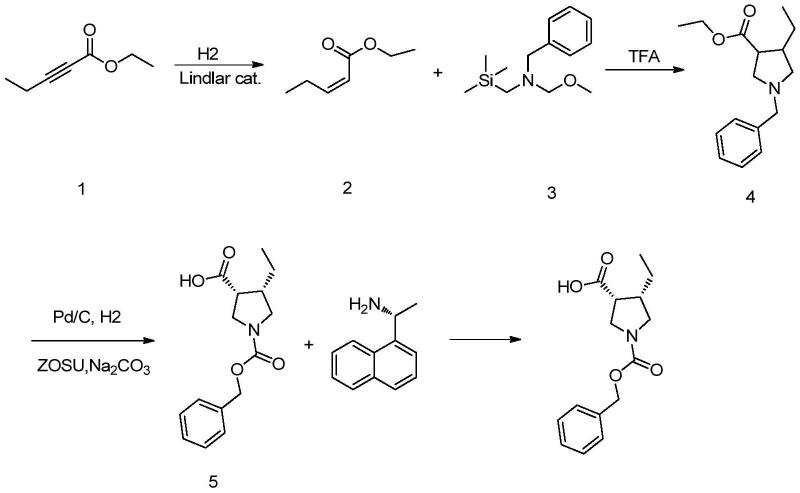
4.wo2011068881报道了乌帕替尼手性中间体(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸的合成的合成方法。该方法以炔基酸酯化合物1为原料,用林德拉催化剂催化氢化还原三键为双键得到化合物2,化合物2与化合物3反应得到消旋体化合物4。化合物4通过化学拆分得到中间体(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸。合成路线如下:
[0005][0006]
该工艺中使用的原料2-戊炔酸乙酯市场不能工业化得到,且工艺报道成环过程,3位羧基和4位乙基均没有手性选择性,不利于后续的手性拆分和手性纯度提高。
[0007]
wo2017066775报道了乌帕替尼手性中间体(3r,4s)-1-((苄氧羰基)-4-乙基吡咯
烷-3-羧酸的合成的合成方法。该方法以cbz-甘氨酸乙酯为原料,碱性条件下与丙烯酸乙酯反应得到化合物1,化合物1与三氟甲磺酸酐反应得到otf取代的中间体2。中间体2与乙基硼酸碱性条件,钯催化催化下偶联反应得到中间体3。中间体3碱性条件水解后的中间体4,中坚体4手性催化剂催化还原和二环己基胺成盐提高手性纯度,最后得到光学纯度合格的(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸。合成路线如下:
[0008][0009]
该合成路线中,使用的起始原料丙烯酸乙酯在2017年10月27日,世界卫生组织国际癌症研究机构公布的致癌物清单初步整理参考,丙烯酸乙酯在2b类致癌物。合成中间体2时,为了活化中间体1的羟基,使用三氟磺酸酐,三氟磺酸酐对水体有很大的污染,对环境和人体极其不友好。合成中间体3时,pd催化剂参与的偶联反应,该步反应需要严格的氮气保护,反应条件较为苛刻,不利于工业化生产。
[0010]
cn111217819报道乌帕替尼手性中间体(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸的合成的合成方法。该方法以中间体1为原料,通过三氯化磷氯代得到中间体1。中间体1与乙基氯化镁,通过ni催化剂偶联得到中间体2。中间体2经手性催化还原和成盐得到(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸。合成路线如下:
[0011][0012]
该合成工艺中,第一步氯代所用试剂三氯氧磷会产生大量的含磷和酸性的污水,对环境不友好。且反应过程,产生的氯化氢会对双键进行加成反应生产杂质。反应体系ph是强酸性,不利于cbz保护基的稳定性。且乙基氯化镁会与中间体2中的酯基进行反应,生成相应的杂质。
[0013]
专利cn 109369659 b报道了一种的合成路线。该路线以羰基中间体1为起始原料,与格氏试剂反应得到羟基中间体2。而后经硫酸脱水、碱水解得到中间体3。该专利中,研究者通过手性ru金属催化剂氢化直接得到手性羧酸中间体,合成路线如下:
[0014][0015]
从该专利的工艺过程来看,第一步乙基溴化镁对羰基进行亲核加成时,乙基溴化镁还会与分子中的酯基进行亲核加成反应,该步反应选择性极差。
技术实现要素:
[0016]
本发明的目的在于:为了解决现有的乌帕替尼中间体(3r,4s)-1-苄氧羰基-4-乙基吡咯烷-3-羧酸合成工艺中反应选择性较差,反应条件苛刻,目标产物光学纯度不高的技
术问题,本发明提供一种乌帕替尼中间体(3r,4s)-1-苄氧羰基-4-乙基吡咯烷-3-羧酸的合成方法。
[0017]
本发明为了实现上述目的具体采用以下技术方案:
[0018]
乌帕替尼中间体(3r,4s)-1-苄氧羰基-4-乙基吡咯烷-3-羧酸的合成方法,包括以下步骤:
[0019]
(1)1-boc-3-吡咯烷酮与dmf-dma反应生成化合物a;
[0020]
(2)化合物a在氮气保护下与甲基溴化镁反应生成化合物b;在该步骤中,化合物a与甲基溴化镁反应过程中会形成六元环中间态,结构式如下:
[0021][0022]
由于结构的特点,反应体系多余的甲基溴化镁不能与羰基反应,从而提高了合成化合物b反应的选择性,减少甲基溴化镁与酮羰基进行反应的杂质;
[0023]
(3)化合物b通过不对称还原羰基为羟基,得到化合物c;该步骤采用羰基的不对称还原corey-bakshi-shibata还原反应,直接得到3位为s构型的3s手性中心,其手性纯度可达到99%,ee值可达到97%;
[0024]
(4)化合物c通过不对称催化还原双键得到化合物d;
[0025]
(5)化合物d在碱性条件下,先和甲基磺酰氯或者对甲苯磺酰氯反应生产中间产物,结构式如下:
[0026][0027]
然后该中间产物与氰基试剂发生亲核取代反应,构型发生绝对翻转得到化合物e;该步骤中,羟基的取代反应运用sn2反应,3位3s手性中心绝对翻转成3r构型,即由s构型oms基团翻转为r构型氰基,有利于目标产物光学纯度的提高;
[0028]
(6)化合物e先在酸性条件下水解氰基为羧基同时脱去boc保护基得到中间产物,结构式如下:
[0029][0030]
再与z-osu或者z-cl反应得到粗品化合物f;
[0031]
(7)粗品化合物f通过重结晶或者化学拆分得到高手性的乌帕替尼中间体(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸;
[0032]
所述化合物a、化合物b、化合物c、化合物d、化合物e、化合物f的结构式依次为:
[0033][0034]
本发明的合成路线为:
[0035][0036]
进一步地,合成化合物a时,反应温度为60-70度,反应时间为8-12小时,溶剂为四氢呋喃、2-甲基四氢呋喃、甲基叔丁基谜、氯仿和二氯甲烷中任意一种,优选使用四氢呋喃。
[0037]
进一步地,合成化合物a时,溶剂与1-boc-3-吡咯烷酮的体积比为1:5,1-boc-3-吡咯烷酮与dmf-dma的摩尔比为1:1.5。
[0038]
进一步地,合成化合物b时,反应温度为-10到-15度,反应时间为3-4小时,溶剂为
四氢呋喃、乙醚和2-甲基四氢呋喃中任意一种,优选使用四氢呋喃。
[0039]
进一步地,合成化合物b时,溶剂与化合物a的体积比为1:10,化合物a与甲基溴化镁的摩尔比为1:1.5。
[0040]
进一步地,合成化合物c时,不对称还原体系选用s-2-甲基-cbs和硼烷四氢呋喃体系,或者七水合三氯化铈和硼氢化钠体系,其中,s-2-甲基-cbs和硼烷四氢呋喃体系s/r比例接近99:2,而七水合三氯化铈和硼氢化钠体系s:r比例只有76:24,优选使用s-2-甲基-cbs和硼烷四氢呋喃体系。
[0041]
进一步地,合成化合物d时,不对称还原体系为以甲醇作溶剂,三乙胺作碱,(s)-segphosru(oac)2做催化剂。
[0042]
进一步地,合成化合物e时,碱性条件选用的碱为三乙胺、n,n-二异丙基乙胺、碳酸钾、碳酸钠和碳酸氢钠中的任意一种,优选使用三乙胺;溶剂选用二氯甲烷、三氯甲烷、四氢呋喃、2-甲基四氢呋喃和dmf中的任意一种,优选使用四氢呋喃;氰基试剂选用氰化钠、氰化钾和三甲基硅氰中的任意一种,优选使用三甲基硅氰。
[0043]
进一步地,合成化合物f时,水解选用的酸为浓盐酸、溴化氢水溶液或稀硫酸,优选使用浓盐酸;水解反应完全后,需将反应ph调节至7-8,在与z-osu或者z-cl反应过程中不断用碱保持ph在7-8,所用的碱可选用三乙胺,n,n-二异丙基乙胺、碳酸钾和碳酸钠中的任意一种,优选使用碳酸氢钠。
[0044]
进一步地,化学拆分试剂选用r-a-苯乙胺、二环己胺或(r)-1-(1-萘基)乙胺,优选使用(r)-(+)-1-(1-萘基)乙胺,化学拆分选用的溶剂为甲醇、乙醇、异丙醇、乙腈、丙酮中任意一种,优选使用乙腈。
[0045]
本发明中各缩写的含义:
[0046]
dmf-dma:n,n-二甲基甲酰胺二甲基缩醛;
[0047]
(s)-segphosru(oac)2:二乙酰基[(r)-(+)-5,5'-双(二苯基膦)-4,4'-双-1,3-苯并二氧杂环戊烷]钌(ii);
[0048]
(s)-2-甲基-cbs:(s)-2-甲基-cbs-恶唑硼烷;
[0049]
z-osu:n-苄氧羰氧基丁二酰亚胺;
[0050]
z-cl:氯甲酸苄酯。
[0051]
本发明的有益效果如下:
[0052]
1.本发明采用简单易得的1-boc-3-吡咯烷酮为原料,通过6步反应得到目标化合物(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸,反应选择性好,反应条件简单,利于工业化生产,目标产物光学纯度高,ee值可达到99.6%。
[0053]
2.本发明化合物a与甲基溴化镁反应过程中会形成六元环中间态,使反应体系中多余的甲基溴化镁不能与羰基反应,从而提高合成化合物b反应的选择性,减少甲基溴化镁与酮羰基反应生成杂质。
[0054]
3.本发明在合成中间体c的过程,采用羰基的不对称还原corey-bakshi-shibata还原反应,直接得到3位为s构型的3s手性中心,其手性纯度达到99%,ee值达到97%,在后续合成化合物e时,羟基的取代反应运用sn2反应,使3位3s手性中心绝对翻转成3r构型,有利于目标产物光学纯度的提高,最后只需要粗品成盐反应就可以得到ee值大于99%的(3r,4s)-1-((苄氧羰基)-4-乙基吡咯烷-3-羧酸。
附图说明
[0055]
图1是化合物a的1h-nmr图谱;
[0056]
图2是化合物b的1h-nmr图谱;
[0057]
图3是化合物c的1h-nmr图谱;
[0058]
图4是化合物d的1h-nmr图谱;
[0059]
图5是化合物f的1h-nmr图谱。
具体实施方式
[0060]
为使本发明实施例的目的、技术方案和优点更加清楚,下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。
[0061]
以下实施例所用原料均为可在市面上购买的工业原料。
[0062]
实施例1
[0063]
化合物a的合成
[0064]
将100g1-boc-3-吡咯烷酮溶于500ml四氢呋喃中,加入64g dmf-dma。将反应液加热70℃,反应8-10小时。tlc检测反应完全后,减压浓缩除去四氢呋喃,浓缩残余物溶于500ml乙酸乙酯,有机相依次用3m的稀盐酸洗涤,饱和食盐水洗涤,无水硫酸钠干燥,减压除去乙酸乙酯得油状物,油状物用甲基叔丁基醚重结晶的黄色固体98.5g,产率76%。纯度99.3%。
[0065]1h nmr(400mhz,cdcl3)δ7.32-7.28(d,j=16hz,1h),4.59-4.54(d,j=20hz,2h),3.87-3.82(d,j=20hz,2h),3.11(s,6h),1.49(s,9h)。
[0066]
实施例2
[0067]
化合物b的合成
[0068]
将90g化合物a溶于900ml四氢呋喃中,保持温度-20至-15℃,氮气保护下取187ml 3m的甲基溴化镁四氢呋喃溶液,缓慢滴入反应液中。滴加完毕后,保持温度温度-20至-15℃反应3-4小时。tlc检测反应完全后,向反应液中滴入10%的氯化铵水溶液100ml。用乙酸乙酯萃取反应液,有机相一次用饱和食盐水洗涤,无水硫酸钠干燥,减压除去乙酸乙酯得红色油状物,油状物用乙酸乙酯:石油醚1:6(v:v)得混合溶剂柱层析得黄色油状物50.6g,收率64%。hplc纯度98.3%。
[0069]1h nmr(400mhz,cdcl3)δ6.78(s,1h),4.32(s,2h),3.93-3.90(d,j=12hz,2h),1.87-1.85(d,j=8.0hz,3h),1.49(s,9h)。
[0070]
实施例3
[0071]
化合物c的合成
[0072]
取355ml硼烷四氢呋喃溶液,控制反应液温度-30℃,缓慢滴入355mls-2-甲基-cbs甲苯溶液中。30分钟后取化合物b50g溶于250ml四氢呋喃中,氮气保护下,保持温度-30℃缓慢滴入反应液中。控制温度小于-30℃反应0.5至1小时,tlc检测反应完全后,向反应液中缓慢加入500ml 10%氯化铵水溶液溶液和500ml乙酸乙酯,有机相用1n盐酸洗洗涤两次,10%碳酸氢钠洗一次,10%盐水洗一次,干燥,浓缩干得油状物,粗品油状物用乙酸乙酯:石油醚=10:1(v:v)柱层析得油状物31g,收率61%,gc纯度95.6%。手性纯度98.6%。
[0073]1h nmr(400mhz,cdcl3):δ5.72-5.71(br,1h),4.57(s,1h),4.13-4.09(d,j=16hz,1h),3.99-3.95(d,j=16hz,1h),3.62-3.59(m,1h),3.44-3.40(m,1h),1.69-1.68(d,j=4hz,3h),1.50-1.48(d,j=8hz,9h)。
[0074]
实施例4
[0075]
化合物d的合成
[0076]
取化合物c取化合物c100g溶于500ml甲醇中,加入(s)-segphosru(oac)20.3g,三乙胺61.6g,将上述溶液缓慢假如高压釜中,并用氢气置换3次后,保持压力3-4mpa,温度70-75℃下反应2-3h。tlc监测反应完全后,氮气置换3次后,过滤除去催化剂。减压浓缩除去甲醇,加入乙酸乙酯,有机相用10%的食盐水和10%的柠檬酸酸水溶液洗涤,无水硫酸钠干燥,减压浓缩后黄色油状物,粗品油状物用粗品油状物用乙酸乙酯:石油醚=6:1(v:v)柱层析得无色油状物82.7g,收率82%,gc纯度93.6%,手性纯度96.4%。
[0077]1h nmr(400mhz,cdcl3):δ3.54-3.41(m,2h),3.28-3.21(m,1h),2.87-2.83(m,1h),2.04-1.95(m,2h),1.46(s,9h),1.42-1.36(m,2h),0.95-0.91(t,j=16hz,3h)。
[0078]
实施例5
[0079]
化合物e的合成
[0080]
取化合物d 50g溶于200ml二氯甲烷中,加入三乙胺70.5g,控制反应温度小于-5℃,缓慢滴加甲基磺酰氯39.9g。滴加完后,再此温度下反应4-6小时。tlc检测反应完全后,取23三甲基氰硅烷加入反应液中,并继续反应8-10小时。tlc监测反应完全后,加入100ml 5%的次氯酸钠水溶液淬灭反应,分出有机相,有机相再用5%的次氯酸钠水溶液洗涤一次(有机相和水相必须用ki钾试制确认氰化物被破坏完全)。然后有机相依次用3m的稀盐酸,10%的氯化钠水溶液和水洗涤。无水硫酸钠干燥,减压浓缩得油状物。油状物未纯化,直接用于下步反应。
[0081]
粗品化合物f的合成
[0082]
将上述油状物溶于50ml四氢呋喃中,加入浓盐酸100ml,加热回流反应8-10小时,tlc监测反应完全后,将反应温度降至室温,反应用10%的碳酸氢钠水溶液调节ph至8-9,缓慢加入z-osu 57.9g,反应过程用10%的碳酸氢钠水溶液保持ph至8-9,tlc检测反应完全后,水相用乙酸乙酯洗涤一次,用6m的盐酸调节ph至2-3,并用乙酸乙酯萃取水相2-3次,合并有机相,有机相用10%的氯化钠水溶液洗涤一次,无水硫酸钠干燥,浓缩得油状物36g,收率56%。hplc纯度94.6%,手性纯度98.4%。
[0083]
实施例6
[0084]
化合物f的拆分
[0085]
取粗品f 20g溶于100ml乙腈中,搅拌下加入(r)-1-(1-萘基)乙胺12.3g,升温至75-80℃反应2-3小时,缓慢降温至25-30℃反应4小时,过滤,烘干得白色固体,将此白色固体加入到300ml水和300ml乙酸乙酯中用柠檬酸调节ph至2-3,分出有机相,有机相用5%的柠檬酸水溶液洗涤2次,10%的氯化钠水溶液洗涤一次,干燥,浓缩得油状物,油状物用乙酸乙酯和石油醚得混合溶剂重结晶得白色固体23.2g,hplc纯度99.2%,ee值99.6%。收率:86%。
[0086]1h nmr(400mhz,cdcl3)δ11.01(br,1h),7.36-7.29(m,5h),5.19-5.10(m,2h),3.82-3.73(m,1h),3.66-3.52(m,2h),3.33-3.24(m,1h),3.14-3.08(m,1h),2.35-2.34(br,
1h),1.56-1.48(m,1h),1.42-1.37(m,1h),0.97(t,j=16hz,3h).
[0087]
实施例7
[0088]
化合物f的拆分
[0089]
取粗品f 20g溶于100ml乙腈中,搅拌下加入r-α-苯乙胺8.7g,升温至75-80℃反应2-3小时,缓慢降温至25-30℃反应4小时,过滤,烘干得白色固体,将此白色固体加入到300ml水和300ml乙酸乙酯中,用柠檬酸调节ph至2-3,分出有机相,有机相用5%的柠檬酸水溶液洗涤2次,10%的氯化钠水溶液洗涤一次,干燥,浓缩得油状物,油状物用乙酸乙酯和石油醚得混合溶剂重结晶得白色固体14g,hplc纯度98.7%,ee值99.2%,收率70%。
[0090]
实施例8
[0091]
化合物f的拆分
[0092]
取粗品f 20g溶于100ml乙腈中,搅拌下加入二环己胺13.3g,升温至75-80℃反应2-3小时,缓慢降温至25-30℃反应4小时,过滤,烘干得白色固体,将此白色固体加入到300ml水和300ml乙酸乙酯中,用柠檬酸调节ph至2-3,分出有机相,有机相用5%的柠檬酸水溶液洗涤2次,10%的氯化钠水溶液洗涤一次,干燥,浓缩得油状物,油状物用乙酸乙酯和石油醚得混合溶剂重结晶得白色固体17.4g,hplc纯度99.5%,ee值98.7%,收率87%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1