一种氯虫苯甲酰胺原药中杂质的制备及表征方法和用途与流程
本发明属于农药化学,具体涉及一种氯虫苯甲酰胺原药中杂质的制备及表征方法和用途。
背景技术:
1、氯虫苯甲酰胺(chlorantraniliprole,cap)是邻甲酰氨基苯甲酰胺类杀虫剂,化学名称为3-溴-n-[4-氯-2-甲基-6-[(甲氨基)羰基)苯基]-1-(3-氯-2-吡啶基)-1h-吡唑-5-酰胺,结构式为:
2、
3、氯虫苯甲酰胺为鱼尼丁受体作用剂,是邻甲酰氨基苯甲酰胺类杀虫剂中的第一个有效成分,广谱、高效防治果树、蔬菜、大田作物、特种作物和草坪上的咀嚼式口器害虫。除了防治鳞翅目害虫外,氯虫苯甲酰胺在增加用药量的情况下,还可以防治科罗拉多甲虫、叶蝉等,同时,对粉虱具有抑制作用,广泛应用于水稻、大豆、棉花、果蔬等农作物。自2008年氯虫苯甲酰胺上市以来,已在世界100多个国家销售,几乎覆盖了所有主要市场,市场前景广阔。
4、由于氯虫苯甲酰胺的合成路线较多,涉及缩合、胺酯交换、关环、开环、水解、卤代、硝化等不同类型的反应,导致氯虫苯甲酰胺原药中的杂质较复杂,分析难度大。而氯虫苯甲酰胺中的杂质可能对其药性或毒理等产生极大的影响,在食品中可能的残留会对消费者产生危害,又或者引起环境污染。因此,对氯虫苯甲酰胺生产过程中杂质的定性和定量分析对氯虫苯甲酰胺的质量控制至关重要。
5、然而,目前市场上高纯度的氯虫苯甲酰胺杂质的对照品供应极少,部分杂质对照品市场无销售,这可能与氯虫苯甲酰胺原药中杂质具有含量低,有效成分和杂质的化学结构属于刚性结构,极性大、溶解性差等特点有关,导致从原药中获取杂质的难度增大。
6、专利文献cn201711460497.5氯虫苯甲酰胺杂质制备工艺和cn202210087659.x一种氯虫苯甲酰胺中间体杂质及其制备方法报道了氯虫苯甲酰胺杂质的合成方法,未涉及从氯虫苯甲酰胺原药中直接分离出杂质的方法。总之,现实情况是,仍然需要开发出简便快速、收率好、纯度高的氯虫苯甲酰胺杂质的分离制备和纯化方法。
7、薄层制备色谱、分析制备色谱是制备毫克级对照品的常用方法。本发明通过打浆、薄层制备色谱和分析制备色谱等分离提纯方式的综合运用,能方便快捷、低成本地从氯虫苯甲酰胺原药中制备氯虫苯甲酰胺原药中的杂质m198、杂质m224、杂质276、杂质m451和杂质m564,且本发明的方法制备的杂质经表征,其纯度能在氯虫苯甲酰胺质量控制中作为杂质对照品。
技术实现思路
1、本发明的第一目的是克服目前市场上高纯度的氯虫苯甲酰胺杂质对照品供应少,且从氯虫苯甲酰胺原药中获取杂质难度较大等问题,提供一种操作简便、杂质收率高的氯虫苯甲酰胺原药中杂质的制备方法。
2、本发明的第二目的是提供一种本发明第一目的制备所得的氯虫苯甲酰胺杂质的表征方法及其在氯虫苯甲酰胺质量控制中作为杂质对照品的应用。
3、为达到本发明的第一目的,本发明提供的技术方案是:
4、一种氯虫苯甲酰胺原药中杂质的制备方法,所述杂质为杂质m198、杂质m224、杂质276、杂质m451和杂质m564,结构式为:
5、
6、制备方法包含如下步骤:
7、(1)氯虫苯甲酰胺原药溶液的制备:将含有杂质m198和/或杂质m224和/或杂质276和/或杂质m451和/或杂质m564的氯虫苯甲酰胺原药用溶剂溶解,通过磁力搅拌打浆或超声处理,得氯虫苯甲酰胺原药溶液;
8、(2)杂质的制备:将步骤(1)中的原药溶液利用薄层制备色谱和/或分析型制备色谱进行制备和纯化。
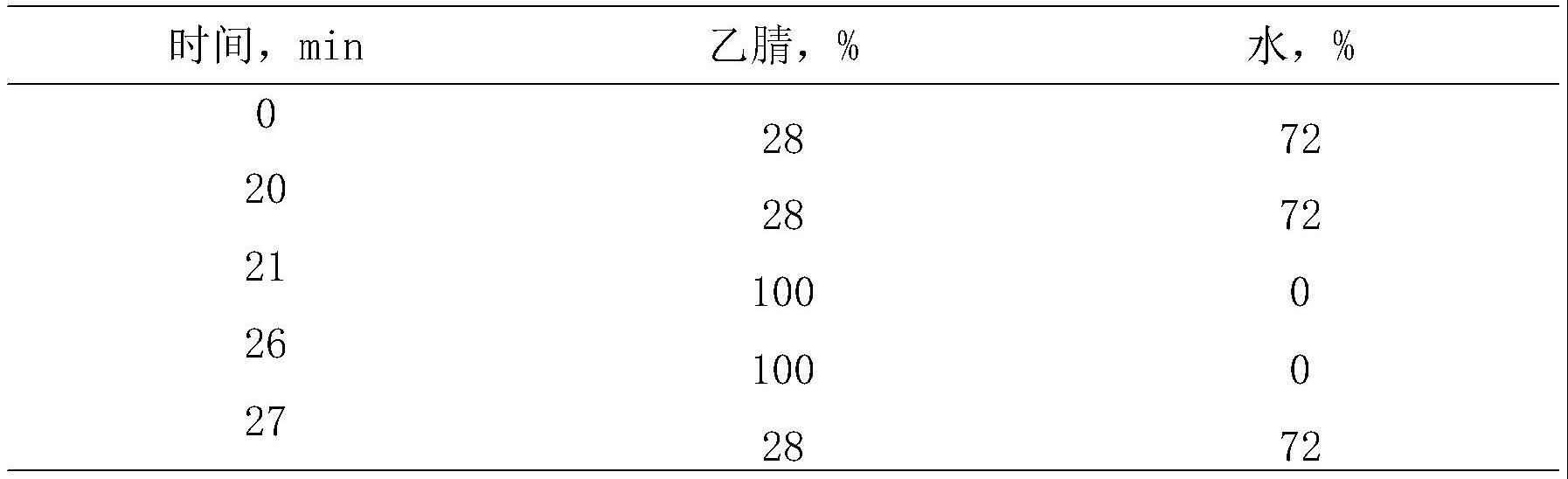
9、步骤(1)中,溶剂为n,n-二甲基甲酰胺或四氢呋喃。
10、步骤(2)中,薄层制备色谱的展开剂为石油醚/乙酸乙酯混合溶剂或二氯甲烷/甲醇混合溶剂或二氯甲烷。
11、步骤(2)中,分析制备色谱的色谱条件为:色谱柱,zorbas sb-c18(250mm×4.6mm,0.5μm);以水、乙腈为流动相,检测波长为254nm,流速为1.0ml/min,柱温为30℃,运行时间为45min,进样量为10~15μl,按洗脱程序进行洗脱。
12、进一步的,当杂质为杂质m564时,制备方法包括如下步骤:
13、(a1)氯虫苯甲酰胺原药溶液的制备:取10g氯虫苯甲酰胺原料置于250ml圆底烧瓶中,加入100ml四氢呋喃,磁力搅拌10分钟,用布氏漏斗抽滤,滤液减压浓缩得10ml浓缩液;
14、(a2)杂质m564的制备和纯化:取足够的硅胶板(20cm*20cm,gf254),将步骤(a1)的浓缩液均匀涂抹于距离硅胶板底部边缘一厘米处,晾干;将硅胶板置于层析缸中,以石油醚/乙酸乙酯=5:1的混合溶剂展开,取出,晾干,刮下硅胶板上的色带,将硅胶色带置于含有20ml二氯甲烷/甲醇=10:1混合溶剂的三角瓶中,超声10分钟,用六孔漏斗抽滤,并用50ml二氯甲烷/甲醇=10:1分三次洗涤硅胶,收集滤液,减压浓缩后即得杂质m564。
15、进一步的,当杂质为杂质m198和m224时,制备方法包括如下步骤:
16、(b1)氯虫苯甲酰胺原药溶液的制备:取10g氯虫苯甲酰胺原料置于250ml圆底烧瓶中,加入100ml四氢呋喃,磁力搅拌10分钟,用布氏漏斗抽滤,滤液减压浓缩至20ml浓缩液;
17、(b2)杂质m198和m224粗品的制备:取足够的硅胶板(规格:20cm*20cm,gf254),将步骤(b1)浓缩液均匀涂抹于距离硅胶板底部边缘一厘米处,晾干;将硅胶板置于层析缸中,以二氯甲烷为展开剂展开后,取出,晾干,分别刮下硅胶板上的色带,将硅胶色带置于含有20ml二氯甲烷/甲醇=10:1混合溶剂的三角瓶中,超声10分钟,用六孔漏斗抽滤,并用50ml二氯甲烷/甲醇=10:1分三次洗涤硅胶,收集滤液,分别取不同色带进行色谱分析,确定目标色带,将目标色带减压去除溶剂后得杂质m198和m224粗品;
18、(b3)杂质m198和m224粗品的纯化:将粗品溶解于5ml四氢呋喃,按杂质m198、m224分析型制备色谱操作条件进行制备,馏分按表4所示的程序进行收集;所得馏分转移至250ml圆底烧瓶,减压旋蒸得杂质纯品m198和粗品m224;将粗品m224按相同的程序进行分析型制备液相二次纯化得纯品m224;
19、杂质m198、m224分析型制备色谱条件为:色谱柱为sb-c18(柱长为250mm,内径为4.6mm,粒径为0.5μm);以水、乙腈为流动相,柱温为30℃;流速为1.00ml/min;检测波长为254nm,进样量15μl,洗脱程序如下:
20、
21、表4:
22、
23、进一步的,当杂质为杂质m276时,制备方法包括如下步骤:
24、(c1)氯虫苯甲酰胺原药溶液的制备:取氯虫苯甲酰胺原药约0.1g,置于100ml容量瓶中,加入10ml n,n-二甲基甲酰胺溶解,超声10分钟后,用乙腈定容至刻度,备用;
25、(c2)杂质m276的制备和纯化:将步骤(c1)所得氯虫苯甲酰胺原药溶液按杂质m276的分析型制备色谱操作条件进行制备,馏分按表5所示的程序进行收集;所得馏分转移至250ml圆底烧瓶,减压旋蒸得杂质纯品m276;
26、其中,杂质m276分析型制备色谱操作条件为:
27、色谱柱为sb-c18(柱长为250mm,内径为4.6mm,粒径为0.5μm);以水、乙腈为流动相,柱温为30℃,流速为1.00ml/min,检测波长为254nm,进样量15μl,洗脱程序如下:
28、
29、表5:
30、
31、进一步的,当杂质为杂质m451时,制备方法包括如下步骤:
32、(d1)氯虫苯甲酰胺原药溶液的制备:取10g氯虫苯甲酰胺原料置于250ml圆底烧瓶中,加入100ml二氯甲烷/甲醇=10:1的混合溶剂,磁力搅拌10分钟,用布氏漏斗抽滤,滤液减压浓缩的10ml浓缩液;
33、(d2)杂质m451薄层制备色谱标样的制备:
34、(d2-1)供试品溶液制备:取约0.1g氯虫苯甲酰胺原药,置于5ml试管中,加入2mln,n-二甲基甲酰胺溶解,超声10分钟,取上清液,过孔径约为0.45μm的有机膜,即得;
35、(d2-2)标样的制备:将步骤(d2-1)所得供试品溶液按杂质m451薄层制备色谱标样色谱操作条件进行制备,馏分按表6所示的程序进行收集;所得馏分转移至250ml圆底烧瓶,减压旋蒸得杂质杂质m451薄层制备色谱标样;
36、其中:杂质451分析型制备色谱条件为:
37、以sb-c18(柱长为250mm,内径为4.6mm,粒径为0.5μm)为色谱柱;以水、乙腈为流动相,柱温为30℃,流速为1.00ml/min,检测波长为254nm,进样量15μl,按如下程序进行洗脱;
38、
39、表6:
40、
41、(d3)杂质m451的制备和纯化:
42、取足够的硅胶板(20cm*20cm,gf254),将步骤(d1)的浓缩液均匀涂抹于距离硅胶板底部边缘一厘米处;并点上步骤(d2)制备所得的杂质451标样,晾干;将硅胶板置于适宜层析缸中,以二氯甲烷/甲醇=10:1的混合溶剂展开,取出,晾干,刮下每一块硅胶板上rf值与标样相同的硅胶色带,合并,将硅胶色带置于含有20ml二氯甲烷/甲醇=10:1混合溶剂的三角瓶中,超声10分钟,用六孔漏斗抽滤,并用50ml二氯甲烷/甲醇=10:1分三次洗涤硅胶,收集滤液,减压去除溶剂后得纯品杂质m451。
43、为达到本发明的第二目的,本发明提供的技术方案是:
44、一种本发明的制备方法制备所得杂质的表征方法,包括如下步骤:
45、ⅰ、供试品溶液的制备:分别取适量本发明的制备方法制备所得的杂质m198、杂质m224、杂质m276、杂质m451和杂质m564,用乙腈溶解即得待测供试品溶液;
46、ⅱ、测定:设置液相色谱和质谱的操作条件,分别对步骤ⅰ所述供试品溶液进行测定,记录色谱图,通过测定结果对供试品进行定性定量分析;
47、液相色谱的操作条件为:
48、色谱柱为sb-c18(柱长为250mm,内径为4.6mm,粒径为0.5μm);以水/乙腈=20/80为流动相,进样量为10μl;流速为1.0ml/min;波长为254nm;柱温为30℃,运行时间为45min;
49、质谱的操作条件为:
50、离子源:esi;
51、采集模式:m198、m276、m451、m564为正离子模式;m224为负离子模式;
52、扫描模式:全扫描;
53、扫描范围:m/z=50~500;
54、碰撞解离电压:70v;
55、干燥气温度:300℃;
56、干燥气流速:7l/min;
57、雾化器压力:15psi;
58、毛细管电压:4000v。
59、本发明制备所得的一种或多种氯虫苯甲酰胺杂质经表征后在氯虫苯甲酰胺质量控制中作为杂质对照品的应用。
60、本发明的有益效果
61、1、本发明通过磁力搅拌打浆、超声、薄层制备色谱和分析制备色谱等分离提纯方式的综合运用,从氯虫苯甲酰胺原药中制备了氯虫杂质m198、杂质m224、杂质276、杂质m451和杂质m564,本发明的方法克服了由于氯虫苯甲酰胺原药中杂质具有含量低,有效成分和杂质的化学结构属于刚性结构,极性大、溶解性差等特点,导致从原药中获取杂质的难度增大的问题,为氯虫苯甲酰胺原药中杂质的制备提供了一种可靠,操作简便快速、成品收率好,纯度高、低成本的制备方法。
62、2、本发明制备所得氯虫苯甲酰胺杂质m198、m224、m276、m451、m564经液相色谱表征,其纯度结果及峰纯度结果显示,每个杂质色谱图只有一个主要的色谱峰,并且峰纯度纯度因子均大于大于设定阈值990.000,说明色谱峰无杂质干扰,成品纯度高。经质谱表征,本发明的制备方法所制得的杂质的质谱结果与杂质m198、m224、m276、m451、m564的结构相符合,能作为氯虫苯甲酰胺质量控制中杂质的对照品使用。
- 还没有人留言评论。精彩留言会获得点赞!