一种α-(1,4)(1,6)-葡聚糖及其制备方法与应用
一种
α-(1,4)(1,6)-葡聚糖及其制备方法与应用
技术领域
1.本发明属于天然多糖技术领域,尤其涉及一种α-(1,4)(1,6)-葡聚糖及其制备方法与应用。
背景技术:
2.植物多糖具有重要的生物学活性,尤其是中药多糖,因其毒副作用小、生物兼容性好,已被广泛用于天然药物和保健食品的开发。目前,以糖类为基础的药物研究日益受到重视并逐渐成为国内外医药界的前沿课题,已有超过50种多糖药物正在进行临床试验,中药来源的多糖物质具有潜在广泛的应用前景。
3.天麻是兰科植物天麻gastrodia elata blume的干燥块茎,性平,味甘,归肝经。天麻作为一种重要的传统中药,始载于《神农本草经》并被列为上品。《中国药典》记载其功效为息风止痉、平抑肝阳、祛风通络,主治小儿惊风、癫痫抽搐、破伤风、头痛眩晕、手足不遂、肢体麻木、风湿痹痛等。它自古以来就被广泛应用于临床治疗,其现代药理作用可归结为“三抗、三镇、一补”,即抗癫痫、抗惊厥、抗风湿,镇静、镇痉、镇痛,补虚,在2021年天麻被正式纳入了国家药食同源名录。
4.天麻多糖(gep)是天麻药效的重要组成部分,药理研究表明这类大分子物质具有抗氧化、抗衰老、免疫调节、神经保护、抑制神经炎症、抑制动脉粥样硬化、抗高血压、改善血脂、抗病毒、抗肿瘤、抗癌、调节肠道菌等多种生物活性。根据文献报道,目前已从天麻中分离纯化到约13种多糖和2种大分子线性葡聚糖,其中仅有7种推测出基本骨架为α-1,4-葡聚糖和α-1,4,6-葡聚糖,少量含有3位取代分支结构。
5.本发明希望针对天麻多糖(gep)展开进一步开发和研究,以期得到新的天麻多糖种类,从而更好利用天麻这一中药资源。
技术实现要素:
6.本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明提出一种α-(1,4)(1,6)-葡聚糖及其制备方法与应用。抗氧化活性筛选结果表明,所述α-(1,4)(1,6)-葡聚糖在20mg/ml浓度下,dpph自由基清除率(drsr)可达到86.24%;在5mg/ml浓度下,羟自由基清除率(hrsr)可达到37.16%,具有较好的抗氧化能力。
7.本发明提供一种α-(1,4)(1,6)-葡聚糖(命名为gep2-6),其相对分子量为2300-3300kda,结构式如下所示:
[0008][0009]
其中,x、y和n为正整数,且x+y=47。
[0010]
将gep2-6进行tfa酸水解-pmp衍生化,再运用液相色谱-质谱法(lc-ms)分析,通过与单糖和寡糖标准品的比对发现,gep2-6主要由葡萄糖(glc)组成,其二糖连接有glc-(1
→
4)-glc和glc-(1
→
6)-glc;再经完全甲基化、水解、还原、乙酰化等反应,运用气相色谱-质谱法(gc-ms)分析其糖苷键连接类型与比例,结果表明gep2-6含有1,4和1,6连接的葡萄糖
残基。结合红外(ft-ir)和核磁(nmr)解析结果显示,明确了所述多糖组分gep2-6为α-(1,4)(1,6)-葡聚糖。
[0011]
优选地,所述α-(1,4)(1,6)-葡聚糖的相对分子量为2500-2900kda。
[0012]
本发明还提供了上述α-(1,4)(1,6)-葡聚糖的制备方法,包括以下步骤:
[0013]
(1)将干燥的天麻药材粉碎、过筛,制成天麻粗粉;
[0014]
(2)将所述天麻粗粉经脱脂、水提、醇沉、洗涤,即得天麻粗多糖;
[0015]
(3)将所述天麻粗多糖脱蛋白后干燥,即得粗多糖粉末;
[0016]
(4)将所述粗多糖粉末用阴离子交换柱进行分离纯化,透析,即得α-(1,4)(1,6)-葡聚糖。
[0017]
优选地,步骤(1)中所述过筛为过10目筛(药典一号筛)且不过80目筛(药典五号筛)。
[0018]
优选地,步骤(2)中采用75%-95%乙醇加热回流进行所述脱脂。
[0019]
更优选地,步骤(2)所述脱脂时的料液比(即天麻粗粉:乙醇,单位为g/ml)为1∶(1-10)。
[0020]
优选地,步骤(2)中采用加热回流进行所述水提,所述水提的料液比为1:(10-20)。
[0021]
优选地,步骤(2)中采用乙醇和丙酮进行所述洗涤。
[0022]
优选地,步骤(3)中采用sevag试剂(氯仿∶正丁醇=4∶1,v/v)进行所述脱蛋白。
[0023]
优选地,步骤(4)中所述阴离子交换柱为deae-52纤维素柱。
[0024]
本发明还提供了上述α-(1,4)(1,6)-葡聚糖在制备抗氧化剂中的应用。试验表明,α-(1,4)(1,6)-葡聚糖在20mg/ml浓度下,dpph自由基清除率(drsr)可达到86.24%;在5mg/ml浓度下,羟自由基清除率(hrsr)可达到37.16%,具有较好的抗氧化能力。
[0025]
本发明还提供了一种抗氧化剂,包含上述α-(1,4)(1,6)-葡聚糖。
[0026]
相对于现有技术,本发明的有益效果如下:
[0027]
本发明提出一种新的天麻多糖(α-(1,4)(1,6)-葡聚糖),该在体外有效清除dpph自由基和羟自由基,具有较好的抗氧化能力。同时,本发明中α-(1,4)(1,6)-葡聚糖的制备方法的产率高,工艺简单,且获得的α-(1,4)(1,6)-葡聚糖纯度高、活性好、易溶于水。
附图说明
[0028]
图1为实施例2中hpsec-elsd法建立葡聚糖在shodex ohpak sb-806-804hq色谱柱上的标准曲线(n=3);
[0029]
图2为实施例2中gep2-6纯度与相对分子量测定色谱图;
[0030]
图3为实施例2中uhplc-q-tof/ms法分析gep2-6的单糖和寡糖组成(a为戊糖,b为脱氧己糖,c为氨基糖,d为己糖,e为糖醛酸,f为己二糖);
[0031]
图4为实施例2中gc-ms法分析gep2-6的糖苷键连接类型;
[0032]
图5为实施例2中gep2-6的红外(ft-ir)扫描光谱;
[0033]
图6为实施例2中gep2-6的核磁(nmr)图谱(a为1h,b为
13
c,c为dept 135,d为hh-cosy,e为hsqc,f为hmbc);
[0034]
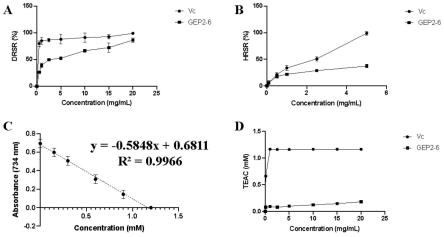
图7为实施例3中gep2-6的抗氧化活性测定结果(n=3,a为dpph自由基清除率,b为羟自由基清除率,c为trolox标准曲线,d为abts法总抗氧化能力检测)。
具体实施方式
[0035]
为了让本领域技术人员更加清楚明白本发明所述技术方案,现列举以下实施例进行说明。需要指出的是,以下实施例仅为本发明的优选实施例,对本发明要求的保护范围不构成限制作用,任何未违背本发明的精神实质和原理下所做出的修改、替代、组合,均包含在本发明的保护范围内。
[0036]
以下实施例中所用的原料、试剂或装置如无特殊说明,均可从常规商业途径得到,或者可以通过现有已知方法得到。
[0037]
实施例1:天麻多糖(gep2-6)的制备
[0038]
本实施例提供一种天麻多糖(命名为gep2-6),其制备方法包括以下步骤:
[0039]
1)将产自四川省广元市的干燥片状的天麻药材粉碎,过药典一号筛且不过药典五号筛收集天麻粗粉。
[0040]
2)称取1468.4g天麻粗粉置多功能提取浓缩机中,先加入7l 95%乙醇浸泡完全,70℃加热回流提取3次、每次1h;抽滤,弃去滤液,将滤渣在室温下放置使其自然干燥,即得脱脂后的天麻粗粉;再加入15l纯水浸泡完全,100℃加热回流提取3次、每次1h,抽滤,合并三次滤液,50℃减压浓缩至1l;再加入3l无水乙醇快速搅拌,4℃静置12h,弃去上清液,用乙醇、丙酮各洗涤黄色粘稠状的沉淀三次。
[0041]
3)将所得沉淀置真空冷冻干燥器中至完全干燥,即得291.33g天麻粗多糖,与原药材相比其产率为19.84%。称取20.355g天麻粗多糖并加入200ml纯水,加热搅拌至完全溶解,sevag法脱蛋白,即用50ml sevag试剂(氯仿∶正丁醇=4∶1,v/v)萃取粗多糖水溶液,剧烈震荡10min,静置30min,去除下层试剂层和中间层变性蛋白,取上层水溶液,重复此操作7次至无白色絮状物出现;将粗多糖水溶液冷冻干燥,即得10.188g脱蛋白后的淡黄色的天麻粗多糖。
[0042]
紫外(uv)检测:称取少量脱蛋白后的天麻粗多糖配制成20mg/ml的水溶液,紫外扫描光谱显示其在200nm处有特征峰,在260和280nm处无吸收峰,说明已几乎不含核酸和蛋白质等杂质。
[0043]
多糖含量测定:采用苯酚-硫酸显色法建立不同浓度的葡萄糖标准曲线(y=0.3814x-0.0141,r2=0.9983),即吸取40μl样品,加入40μl现配的5%苯酚溶液,再迅速加入160μl浓硫酸后,混匀,70℃加热10min,冷却至室温,使用酶标仪在490nm下检测吸光度。根据线性回归方程公式,可计算出天麻粗多糖在脱蛋白前的多糖含量仅为55.79%,而脱蛋白后多糖含量上升至86.73%。
[0044]
4)制备deae-52纤维素柱进一步分离纯化天麻粗多糖,具体实验过程如下:
[0045]
a.装柱:将100g deae-52纤维素填料中加入约4l纯水,充分搅拌并浸泡约24h,弃去漂浮的杂质,再连续倾入至底部塞有少量棉花、内径约为5cm的玻璃柱内,填料自然沉降高度约为40cm;
[0046]
b.活化:配制等柱体积的0.5mol/l naoh溶液浸润填料约2h,用纯水连续冲洗约5个柱体积至流出液呈中性后,再加入等柱体积的0.5mol/l hcl溶液浸润填料约2h,同法冲洗至中性后,再加入等柱体积的0.5mol/l naoh溶液浸润填料约2h,同法冲洗至中性;
[0047]
c.上样:称取1.986g粗多糖粉末并加入20ml纯水,加热搅拌至完全溶解,滴加样品水溶液至填料表面的滤纸上;
[0048]
d.洗脱:控制流出液流速和加入洗脱剂的蠕动泵流速均为0.7ml/min,先用纯水洗脱2个柱体积,再用0.1mol/l nacl洗脱3个柱体积,收集前700ml的流出液,40℃减压浓缩至约10ml;
[0049]
e.透析:将浓缩的0.1mol/l nacl洗脱液倒入截留分子量为1000da的透析袋内,完全浸没于大量流动纯水中,磁力搅拌透析约24h至透析袋不再膨胀,冷冻干燥得732.1mg白色棉絮状的多糖组分gep2-6,与原药材相比其产率为3.66%。
[0050]
实施例2:多糖的结构表征
[0051]
1)高分子量多糖的纯度与相对分子量测定
[0052]
样品处理:将不同分子量(4.66、12.6、25、63.3、126、556、2000kda)的葡聚糖标准品和实施例1制得的天麻多糖gep2-6分别用超纯水配制成5mg/ml的溶液,建立标准曲线(如图1所示,n=3)。
[0053]
高效排阻液相色谱(hpsec)条件:使用agilent 1260infinity hplc系统和elsd检测器,由聚合物基质水溶性sec色谱柱shodex ohpak sb-806hq和shodex ohpak sb-804hq串联分离,理论塔板数≥16000,排阻限分子量范围可达5-20000kda;柱温稳定保持在26℃,超纯水等度洗脱,流速为1ml/min,进样量为30μl,检测时间为25min。
[0054]
如图2所示,天麻多糖gep2-6的纯度为98.034%,根据图1中的拟合曲线公式计算其平均相对分子量为2722.68kda。
[0055]
2)单糖和寡糖组成分析
[0056]
样品处理:先称取1mg gep2-6加入1ml 4mol/l三氟乙酸(tfa)完全溶解后,120℃酸水解2h,吸取10μl加入50μl甲醇,n2吹干,如此重复醇洗5次;再将吹干的水解样品与原浓度为1mg/ml的单糖(戊糖:核酮糖、核糖、木糖、阿拉伯糖,脱氧己糖:鼠李糖、岩藻糖,氨基糖:氨基葡萄糖、氨基半乳糖,己糖:果糖、甘露糖、阿洛糖、葡萄糖、半乳糖,糖醛酸:葡萄糖醛酸、半乳糖醛酸)和二糖(曲二糖glc-(1
→
2)-glc、麦芽糖glc-(1
→
4)-glc、异麦芽糖glc-(1
→
6)-glc、纤维二糖glc-(1
→
4)-glc)标准品同时进行1-苯基-3-甲基-5-吡唑啉酮(pmp)衍生化,即分别加入20μl氨水和20μl 0.6mol/l pmp/甲醇溶液,60℃反应40min,n2吹干,水-氯仿萃取上层水溶液并稀释一定倍数。
[0057]
液相色谱-质谱(lc-ms)条件:使用agilent 6550uhplc-q-tof/ms系统,色谱柱为agilent polaris 3c18-ether,柱温为30℃,流动相a(5%乙腈水溶液加25mm醋酸铵,氢氧化铵调节ph值至8.4)和流动相b(50%乙腈水溶液)进行梯度洗脱(0-0.5min,15%b;0.5-7min,15%-35%b;7-11min,35%-95%b;11-13.9min,95%b;13.9-14min,95%~15%b),流速为0.3ml/min,进样量为1μl。质谱数据采集为正模式,m/z 922.0098(c
18h18f24
n3o6p3)作为校正以获得准确质量。
[0058]
如图3所示,根据提取离子色谱图并通过与标准品的比对发现,gep2-6主要由葡萄糖(glc)组成,其二糖连接有glc-(1
→
4)-glc和glc-(1
→
6)-glc。
[0059]
3)糖苷键连接分析
[0060]
样品处理:先称取1mg天麻多糖gep2-6加入1ml 120mg/ml naoh/dmso混悬液,充入n2后密封,冰浴超声30min,加入1ml碘甲烷超声1h,水-二氯甲烷萃取三次,将下层二氯甲烷相吹干,重复上述操作两次至达到完全甲基化;再依次经4mol/l tfa水解3h、甲醇洗5次,0.5mol/l nabh4室温还原反应2h、冰醋酸中和、甲醇洗5次,最后用醋酐在120℃经乙酰化反
应1h、水-二氯甲烷萃取三次,将二氯甲烷相n2吹浓缩,高速离心取上清液。
[0061]
气相色谱-质谱(gc-ms)条件:使用agilent 7890a gc-fid/msd系统,色谱柱为db-5ms,氦气流速为1ml/min,进样量为1μl,初始温度为130℃并保持5min,再以3℃/min速率增至250℃并保持5min。
[0062]
如图4所示,根据提取离子色谱图并通过与nist数据库的比对发现,天麻多糖gep2-6含有1,4和1,6连接的葡萄糖残基,经糖苷键连接分析实验测得上述两种葡萄糖残基的摩尔比约为31.18∶1.32。
[0063]
4)红外(ft-ir)分析
[0064]
红外扫描光谱见图5显示,3379cm-1
宽峰为o-h伸缩振动,2940cm-1
弱峰为c-h伸缩振动,1026-1150cm-1
为c-o-c和c-o-h的糖环振动信号,1700cm-1
附近没有吸收峰,明确了gep2-6为中性多糖。
[0065]
5)核磁(nmr)分析
[0066]
将80mg天麻多糖gep2-6溶于800μl含0.05%tsp内标的d2o中,使用bruker 600mhz nmr波谱仪测定,如图6所示,由1h、
13
c、dept 135、hh-cosy、hsqc和hmbc谱解析归属信号(ppm,δ)如下:α-d-1,4-glcp:h1/c1(5.42/102.33),h2/c2(3.66/74.21),h3/c3(3.98/76.02),h4/c4(3.67/79.46),h5/c5(3.86/73.86),h6a,b/c6(3.84,3.78/63.11);α-d-1,6-glcp:h1/c1(4.99/101.33),h2/c2(3.62/74.41),h3/c3(3.72/75.38),h4/c4(3.44/72.00),h5/c5(4.06/73.04),h6a,b/c6(3.96,3.88/70.42)。结果表明,gep2-6为α-(1,4)(1,6)-葡聚糖。
[0067]
实施例3:水溶性高分子量α-(1,4)(1,6)-葡聚糖的富集
[0068]
参照实施例1,将步骤3)脱蛋白后的天麻粗多糖用deae-52纤维素柱层析多次纯化,每次上样1-2g,控制流速为0.7ml/min,先用纯水洗脱2个柱体积,再用0.1mol/l nacl洗脱3个柱体积,收集前700-800ml的流出液透析;2mol/lnacl洗脱残留物后,重复上样富集0.1mol/l nacl洗脱部位,即得一种水溶性高分子量α-(1,4)(1,6)-葡聚糖(结果见表1)。
[0069]
表1
[0070][0071]
实施例4:水溶性高分子量α-(1,4)(1,6)-葡聚糖的抗氧化活性筛选试验
[0072]
1)dpph自由基清除率
[0073]
将1,1-二苯基-2-苦基肼自由基(dpph)用无水乙醇配制成0.1mm的dpph溶液,对照孔ac为100μl超纯水加入20μl dpph溶液,对照背景孔a
cb
为100μl超纯水加入20μl无水乙醇;测定孔as为100μl样品加入20μl dpph溶液,测定背景孔a
sb
为100μl样品加入20μl无水乙醇。混匀,室温避光反应30min,使用酶标仪在517nm下检测吸光度。dpph自由基清除率(dpph radical scavenging rate)计算如下:
[0074]
[0075]
2)羟自由基清除率
[0076]
按羟自由基清除能力检测试剂盒(索莱宝,bc1325)说明书操作,空白孔ab为40μl超纯水加入100μl工作液;对照孔ac为20μl超纯水加入100μl工作液再加入20μl试剂四工作液;测定孔as为20μl样品加入100μl工作液再加入20μl试剂四工作液。混匀,37℃避光反应1h,使用酶标仪在536nm下检测吸光度。羟自由基清除率(hydroxyl radical scavenging rate)计算如下:
[0077][0078]
3)abts法总抗氧化能力检测
[0079]
按abts法总抗氧化能力检测试剂盒(碧云天,s0119)说明书操作,对照孔ac为10μl超纯水加入200μl abts工作液,对照背景孔a
cb
为10μl超纯水加入200μl pbs缓冲液;测定孔as为10μl样品加入200μl abts工作液,测定背景孔a
sb
为10μl样品加入200μl pbs缓冲液。混匀,室温避光5min,使用酶标仪在734nm下检测吸光度。根据阳性对照trolox标准曲线公式,可计算出trolox相当的抗氧化能力(trolox-equivalent antioxidant capacity,teac/mm)。
[0080]
如图7所示,与维生素c(vc)相比,天麻多糖gep2-6在20mg/ml浓度下其drsr可达到86.24%,而在5mg/ml浓度下其hrsr可达到37.16%;但gep2-6的abts自由基清除能力teac稍弱。
[0081]
上面结合附图对本技术实施例作了详细说明,但是本技术不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本技术宗旨的前提下作出各种变化。此外,在不冲突的情况下,本技术的实施例及实施例中的特征可以相互组合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1