一种基于白藜芦醇和异香兰素生物基复合环氧树脂及其制备方法
1.本发明属于材料领域,具体涉及一种基于白藜芦醇和异香兰素生物基复合环氧树脂及其制备方法。
背景技术:
2.环氧树脂是目前聚合物材料中应用较多的一种,主要呈现热固性的特征,相对于热塑性的材料而言,热固性树脂材料具有更高的使用温度,而不会出现熔融和熔断现象,在力学性能、热学性能以及耐腐蚀性等方面均发挥较好的功能化作用,以此为基础开发的环氧树脂聚合物材料在涂料和电子密封胶等方面具有较好的应用,因此在工程塑料、工程复合材料和表层防护等方面均具有很好的应用价值。
3.环氧树脂的全球产能在500万吨/年左右,其中基础性的品种仍然是以双酚a类环氧树脂结构为主。全球范围内的涂料和油漆等产业急速发展,意味着环氧树脂的市场消耗量较大。对于双酚a环氧树脂的使用中存在过度依赖和较为显著的安全隐患,最近在生物基替代等方面也逐渐形成了一些相应结构或原料的替代趋势。
4.生物基环氧树脂替代的根本原因不仅在于现有石油化学品的过度开采导致资源紧张问题,更主要的原因之一还在于用于制备双酚a的丙酮和苯酚以及强酸制备工艺等都受石化资源等因素限制。此外,双酚a市场份额巨大,在环氧树脂和聚碳酸酯等聚合物等方面研究占比也较高,但是双酚a类树脂对人体生殖健康存在消极影响,已被美国联邦药物管理局明令禁止用作婴儿配方奶粉的包装材料,其相应的禁用领域也在逐步拓宽。同时,相应的环氧树脂材料具有较好的耐磨性,但是,长期使用过程中产生的颗粒状微粒也进一步使得材料在环境中容易长期累积,从而也会出现可能的二次安全污染。
5.对生物基材料替代的需求以及其在安全性方面的考虑,广泛地发展生物基材料,开发安全性相对较高的生物质环氧树脂,在高性能、高生物安全性等方面以代替石油资源的环氧树脂,在未来的发展过程中具有非常好的前景。
6.此外,传统的双酚a环氧树脂结构,因为在热稳定性方面效果较差,同时燃烧污染程度严重,受热后残碳含量接近于零,因此碳含量较低,然而基于生物基环氧树脂的分解炭化过程制备生物基炭材料的研究目前研究较少。
技术实现要素:
7.本发明所要解决的技术问题是针对现有技术的不足,提供一种基于白藜芦醇和异香兰素生物基复合环氧树脂及其制备方法。
8.为了解决上述技术问题,本发明公开了一种基于白藜芦醇和异香兰素生物基复合环氧树脂的制备方法,包括如下步骤:
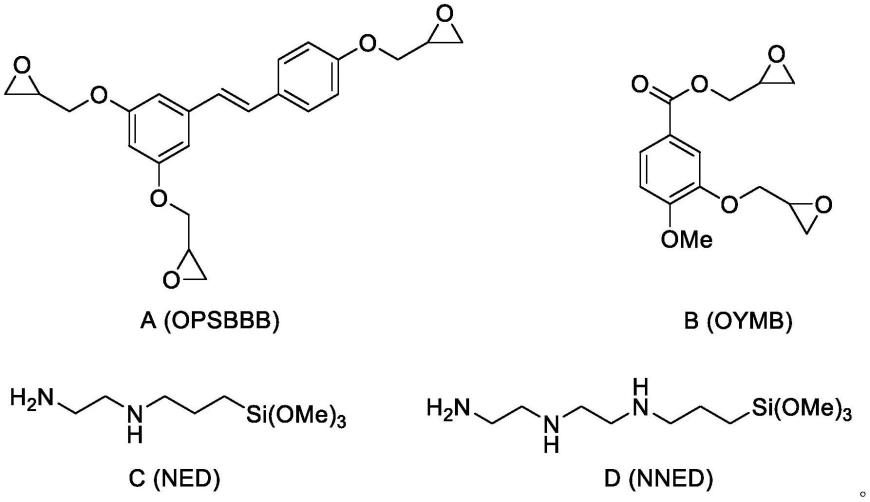
9.(1)将式a所示的化合物与式b所示的化合物进行搅拌混匀,得到双生物基环氧单体复合物;
10.(2)将步骤(1)得到的双生物基环氧单体复合物与固化剂进行混合,熔融,固化,即得;
11.其中,所述的固化剂为式c所示的固化剂和/或式d所示的固化剂;
[0012][0013]
其中,步骤(1)中,所述的式a所示的化合物的制备,具体方法为:在室温下,向反应瓶中依次加入白藜芦醇、三乙基苯基氯化铵和环氧氯丙烷,磁力搅拌加热反应,反应结束后冷却至室温;随后向体系中加入三乙基苯基氯化铵和氢氧化钠水溶液,在室温下搅拌反应,反应结束后萃取反应液,分层,有机相干燥,过滤,浓缩,通过硅胶柱层析纯化,即得。
[0014]
其中,步骤(1)中,所述的式b所示的化合物的制备,具体方法为:向反应瓶中依次加入氢氧化钠、氢氧化钾和去离子水,加热体系,然后向体系中加入异香草醛,加热反应;反应结束后,用磷酸对体系进行酸化,直至室温下ph=2,过滤,固体部分用水冲洗,干燥,得到中间体;随后,依次将得到的中间体、苄基三乙基氯化铵、环氧氯丙烷加入到反应瓶中,加热反应,接着继续向体系中加入苄基三乙基氯化铵和氢氧化钠水溶液,并在室温下继续反应;反应结束后萃取反应液,分层,有机相用水洗涤,分层,有机相干燥,浓缩,通过硅胶柱层析纯化,即得。
[0015]
具体地,步骤(1)中,式a所示的化合物中的环氧基团与式b所示的化合物中的环氧基团的摩尔比为n,0《n《100,优选为0.3≤n≤3。
[0016]
具体地,步骤(2)中,双生物基环氧单体复合物中的环氧基团与固化剂中的nh的摩尔比为2:1~4。
[0017]
其中,所述的双生物基环氧单体复合物中的环氧基团与固化剂中的nh通过c-n键合。
[0018]
具体地,步骤(2)中,所述的熔融,熔融温度为50~75℃。
[0019]
具体地,步骤(2)中,所述的固化,固化温度为65~95℃;所述的固化温度大于等于熔融温度。
[0020]
具体地,步骤(2)中,所述的固化,固化时间为2~5h。
[0021]
上述的制备方法制备得到的基于白藜芦醇和异香兰素生物基复合环氧树脂也在
本发明的保护范围之内。
[0022]
所述的基于白藜芦醇和异香兰素生物基复合环氧树脂在制备生物基炭材料中的应用也在本发明的保护范围之内。
[0023]
有益效果:
[0024]
(1)本发明提供一种基于白藜芦醇、异香兰素的复合型生物基环氧树脂的制备方法,环氧单体的绿色化清洁化程度高,且固化工艺过程温和高效;
[0025]
(2)本发明所提供的一种基于生物基环氧单体制备的材料进一步构建通过氧化的方式构建生物基炭基;
[0026]
(3)本发明中基于氧化的方法制备得到生物基碳含量相对较高,便于进一步拓展对于生物质化炭材料的应用研究。
附图说明
[0027]
下面结合附图和具体实施方式对本发明做更进一步的具体说明,本发明的上述和/或其他方面的优点将会变得更加清楚。
[0028]
图1为式a所示的白藜芦醇生物基环氧树脂单体的合成路线。
[0029]
图2为式a所示的白藜芦醇生物基环氧树脂单体的核磁共振氢谱。
[0030]
图3为式a所示的白藜芦醇生物基环氧树脂单体的核磁共振碳谱。
[0031]
图4为式b所示的异香兰素生物基环氧树脂单体的合成路线。
[0032]
图5为图4中所示中间体e的核磁共振氢谱。
[0033]
图6为图4中所示中间体e的核磁共振碳谱。
[0034]
图7为式b所示的异香兰素生物基环氧树脂单体的核磁共振氢谱。
[0035]
图8为式b所示的异香兰素生物基环氧树脂单体的核磁共振碳谱。
[0036]
图9为实施例9所得聚合物材料的raman谱图;其中,对应的峰积分面积比ig/id=0.328。
[0037]
图10为实施例10所得聚合物材料的raman谱图;其中,对应的峰积分面积比ig/id=0.317。
[0038]
图11为实施例3和4所得聚合物材料的热重谱图。
[0039]
图12为实施例9和10所得聚合物材料的热重谱图。
[0040]
图13为实施例5和6所得聚合物材料的热重谱图。
[0041]
图14为实施例3~12中所得聚合物材料的红外谱图。
具体实施方式
[0042]
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0043]
原料合成部分实施例:
[0044]
实施例1
[0045]
在室温下,向100ml三口反应瓶中依次加入白藜芦醇(1.0g),三乙基苯基氯化铵(0.29g)和环氧氯丙烷(12.0g)并磁力均匀搅拌,得到混合物;将所得混合物在80℃搅拌反应2h,反应结束后将反应液冷却至室温;随后继续向体系中加入三乙基苯基氯化铵(0.29g)
和氢氧化钠水溶液(2.06g,5.0mol/l),并在室温下搅拌反应直到反应完成,反应结束后反应液用乙酸乙酯萃取,分层,有机相用无水硫酸镁干燥,过滤,滤液浓缩,通过硅胶柱层析纯化得到淡黄色固体产物式a化合物1.6g,收率为92%;式a化合物的合成路线如图1所示,式a化合物的核磁共振氢谱如图2所示,核磁共振碳谱如图3所示。
[0046]
实施例2
[0047]
向500ml烧瓶中依次添加氢氧化钠(32.17g)、氢氧化钾(48.25g)和去离子水(16ml),然后在10min内将混合物加热至160℃,随后向体系中添加异香草醛(96.5g,0.635mol),将反应体系在160℃下持续反应4h;反应结束后,将反应混合物转移到烧杯中,用磷酸对混合物进行酸化,直到室温下ph=2,过滤,将所得沉淀物用水冲洗,最后干燥,得到固体中间体e 92g,产率为86.2%;中间体e的合成路线如图4所示,中间体e的核磁共振氢谱如图5所示,核磁共振碳谱如图6所示。
[0048]
将中间体e(2.52g)、苄基三乙基氯化铵(tebac,0.34g)、环氧氯丙烷(13.8g)添加到烧瓶(250ml)中,并在80℃下通过磁力搅拌使混合物反应4.5h,随后添加另一份tebac(0.34g)和氢氧化钠水溶液(2.4g,5.0mol/l),并在室温下进行反应1.5h;反应结束后,用乙酸乙酯萃取混合物三次,分层,用水洗涤合并的有机相三次,分层,有机相用无水硫酸钠干燥,过滤,滤液浓缩,使用pe/ea作为洗脱液,通过硅胶柱层析纯化混合物,得到白色固体式b化合物3.0g,收率为71.3%;式b化合物的核磁共振氢谱如图7所示,核磁共振碳谱如图8所示。
[0049]
聚合部分实施例:
[0050]
实施例3
[0051]
ospbbb/ned聚合物的制备
[0052]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.1586g,实施例1制备得到),在25℃下计量加入固化剂c(0.1334g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至75℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0053]
实施例4
[0054]
ospbbb/nned聚合物的制备
[0055]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.1586g,实施例1制备得到),25℃下计量加入固化剂d(0.1593g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至70℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0056]
实施例5
[0057]
50%ospbbb/50%oymb/ned聚合物的制备
[0058]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.0793g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.0841g,实施例2制备得到),25℃下计量加入固化剂c(0.1334g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至80℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0059]
实施例6
[0060]
50%ospbbb/50%oymb/nned聚合物的制备
[0061]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.0793g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.0841g,实施例2制备得到),25℃下计量加入固化剂d(0.1593g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至80℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0062]
实施例7
[0063]
75%oymb/25%ospbbb/ned聚合物的制备
[0064]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.0395g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.1261g,实施例2制备得到),25℃下计量加入固化剂c(0.1334g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至90℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0065]
实施例8
[0066]
75%oymb/25%ospbbb/nned聚合物的制备
[0067]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.0395g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.1261g,实施例2制备得到),25℃下计量加入固化剂d(0.1593g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至80℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体而言,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0068]
实施例9
[0069]
oymb/ned聚合物的制备
[0070]
反应瓶中称取式b所示的异香兰素生物基环氧树脂单体(0.109g,实施例2制备得到),25℃下计量加入固化剂c(0.086g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至95℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体,出现较大的吸收,推断为过程中形成的大量羟基导致的。空气下,样品水平45
°
放置,后点火5s,撤离火源,维持环氧树脂自燃烧直至熄灭,后残碳直接用于拉曼测试,经分析规则石墨烯g位于1590cm-1
,不规则的石墨烯结构d位于1356cm-1
处,如图9所示。
[0071]
实施例10
[0072]
oymb/nned聚合物的制备
[0073]
反应瓶中称取式b所示的异香兰素生物基环氧树脂单体(0.109g,实施例2制备得到),25℃下计量加入固化剂d(0.1356g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至85℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体,出现较大的吸收,推断为过程中形成的大量羟基导致的。空气下,样品水平45
°
放置,后点火5s,撤离火源,维持环氧树脂自燃烧直至熄灭,后残碳直接用于拉曼测试,经分析规则石墨烯g位于1590cm-1
,不规则的石墨烯结构d位于1356cm-1
处,如图10所示。
[0074]
实施例11
[0075]
25%oymb/75%ospbbb/ned聚合物的制备
[0076]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.111g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.0392g,实施例2制备得到),25℃下计量加入固化剂c(0.1245g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至80℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧的开环过程,3434cm-1
处相对于原料环氧单体,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0077]
实施例12
[0078]
25%oymb/75%ospbbb/nned聚合物的制备
[0079]
反应瓶中称取式a所示的白藜芦醇生物基环氧树脂单体(0.111g,实施例1制备得到)和式b所示的异香兰素生物基环氧树脂单体(0.0392g,实施例2制备得到),25℃下计量加入固化剂d(0.1486g),升温至大约60℃,样品处于熔融状态,快速搅拌保持物料充分熔融,混合均匀。后逐渐升温至80℃开始固化,后在此温度下维持2h后得到透明的淡黄色聚合物材料。如图14所示,通过对其红外数据的判断,原有环氧底物中环氧乙烷红外峰(860和910cm-1
等强度伸缩振动)消逝,表明环氧树脂的环氧基团与胺基已完全聚合,同时由于环氧
的开环过程,3434cm-1
处相对于原料环氧单体,出现较大的吸收,推断为过程中形成的大量羟基导致的。
[0080]
表1双生物基环氧树脂的热稳定性与700℃度下的残碳含量
[0081][0082]
从表1得知,向oymb/ned体系中增加ospbbb,整体上有利于t
d30
和t
max
以及r
700
的增加,便于提升材料的热稳定性和成炭率;尽管ospbbb/ned体系的成炭性能与oymb/ned相似,但通过有效的复配仍然可以使r
700
提升7%左右(实施例7vs实施例9);另外在热性能方面通过50%的ospbbb的掺入,可以提高oymb/nned的t
max
约40℃,t
d30
提高到400℃以上(实施例6vs实施例10)。
[0083]
表2实施例3~实施例6燃烧碳结构形貌
[0084][0085][0086]
本发明提供了一种基于白藜芦醇和异香兰素生物基复合环氧树脂及其制备方法的思路及方法,具体实现该技术方案的方法和途径很多,以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。本实施例中未明确的各组成部分均可用现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1