一组结肠癌DNA甲基化分子标志物及其在制备用于结肠癌早期诊断试剂盒中的应用
本发明涉及生物检测,具体涉及一组结肠癌的dna甲基化分子标志物及其在制备用于结肠癌诊断试剂盒中的应用。
背景技术:
1、
2、结肠癌疾病的确切原因尚不清楚,然而,一些特定的危险因素与结肠癌高度相关,包括不健康饮食、大量饮酒和吸烟。此外,有结肠癌家族史或有某些遗传性癌症综合征的人患此病的风险很高。细胞中的基因突变是结肠癌最常见的原因之一。由于缺乏有效的诊断方法,结肠癌在早期是不容易被发现的。
3、尽管已经有一些关于结肠癌的分子探索、生物标志物和治疗靶点,这些对治疗和诊断这种疾病有很大的贡献,但由于生物的复杂性和较大的肿瘤个体差异性,目前急需具有更高稳定性和准确性的结肠癌标志物。
4、dna甲基化是一种在基因调控中起重要作用的表观遗传标志物。异常的dna甲基化修饰与许多疾病有关。并且,甲基化标志物比蛋白质标志物更稳定,这明确了癌症特异性甲基化标志物具有巨大的潜力,可用于在临床上准确诊断癌症。但是,目前关于甲基化标志物在结肠癌中的应用较少,大多数找到的甲基化标志物均为特定基因中的启动子或差异甲基化区域,这让检测过程复杂、检测的成本也较高。
技术实现思路
1、本发明的目的在于通过甲基化测序数据和rna-seq测序数据的整合分析,进一步结合机器学习方法,提供一组结肠癌dna甲基化分子标志物及其在制备用于结肠癌早期诊断试剂盒中的应用。本发明选择结肠癌dna甲基化标志物的原则是:尽可能选出最具有标志性的位点,以降低检测的复杂性和实验成本。
2、本发明的目的是通过以下技术方案来实现的,一组结肠癌甲基化分子标志物,所述标志物包括如下30个cpg位点的甲基化:cg06668555、cg06392169、cg18596362、cg13265789、cg21782409、cg04555373、cg01893212、cg04904331、cg12584684、cg04454951、cg04804539、cg20078466、cg09493505、cg05470523、cg19202058、cg00333226、cg15701178、cg01194057、cg01610488、cg07039180、cg04279973、cg06716730、cg18500968、cg03462053、cg05000488、cg14898779、cg14018648、cg19981409、cg05407490和cg0533134。所述30个cpg位点分布在16个不同基因上,包括fam135b、irf4、cbln2、unc5c、nrg1、vwc2、mal、ikzf1、trpa1、prkcb、dusp14、tmprss3、krt6a、stk31、asgr1和nox4;其中9个基因与结肠癌患者的生存率明显相关,分别是nox4、krt6a、dusp14、asgr1、nrg1、trpa1、irf4、prkcb和ikzf1。
3、本发明还提供了一种上述dna甲基化分子标志物在制备用于结肠癌早期诊断试剂盒中的应用。
4、进一步地,以筛选出的30个cpg位点的dna甲基化水平为基础,利用methyltargt测序方法分析肿瘤组织样本、内镜活检样本或血浆/血清样本,采用支持向量机(svm)方法构建结肠癌诊断的数学模型,使用roc曲线及曲线下面积(auc)来评价筛选效果。
5、本发明还提供一种上述dna甲基化分子标志物在制备结肠癌预后试剂盒中的应用,所述分子标志物的30个cpg位点位于9个基因上,所述9个基因分别为nox4、krt6a、dusp14、asgr1、nrg1、trpa1、irf4、prkcb和ikzf1。
6、本发明的结肠癌甲基化分子标志物通过以下方法获取:
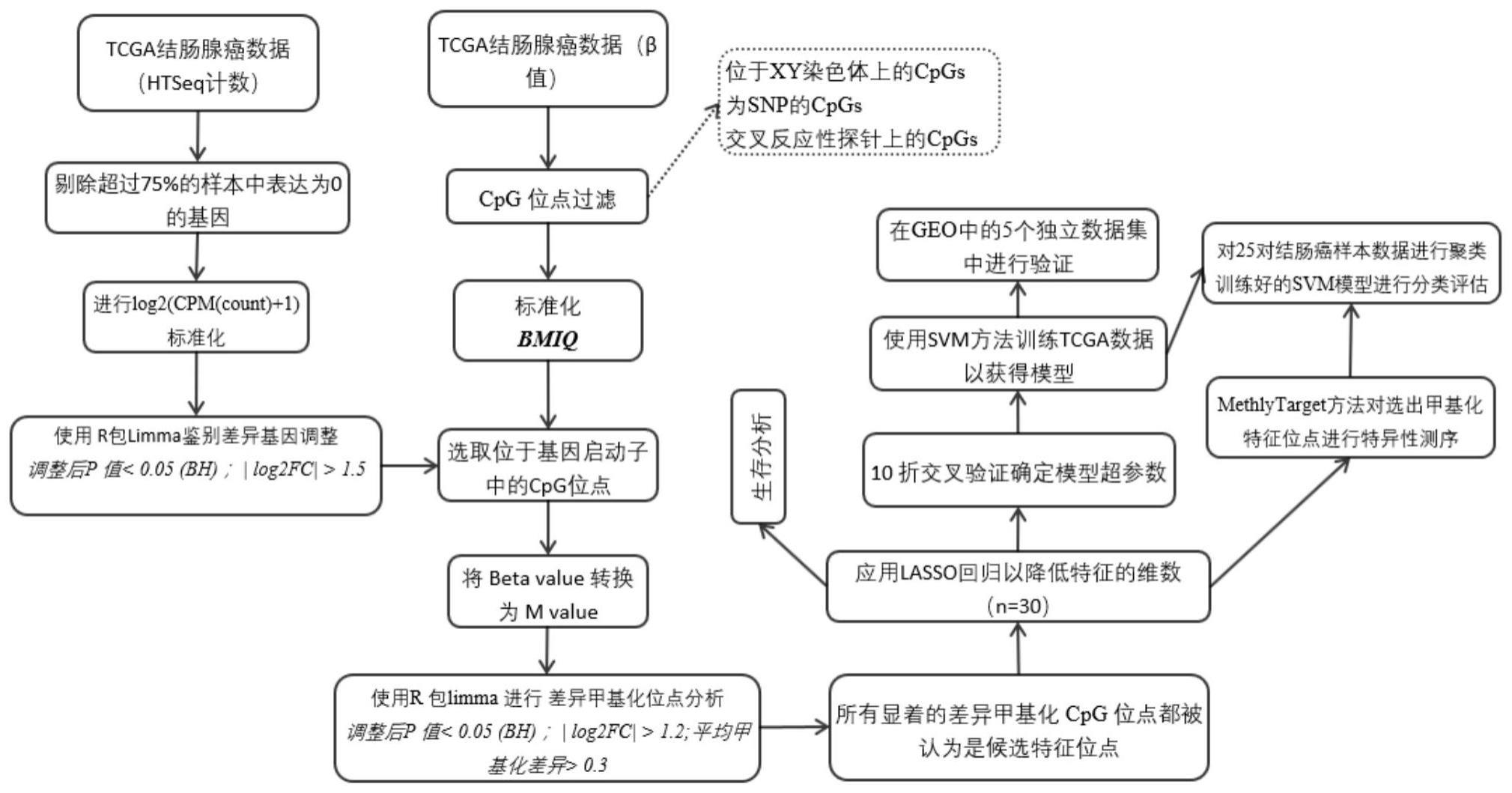
7、(1)甲基化和基因表达数据的获取:从tcga下载结肠癌(coad)的基因表达和dna甲基化的数据集。其中,共有295个结肠癌病人同时具有甲基化和基因表达这两种数据,选择这些病人的样本数据用于后续分析。对于这295个病人来说,他们的基因表达数据集中共有包含346个样本,包括317个结肠癌和29个癌旁样本,而甲基化的数据集总共包含352个样本,包括314个结肠癌和38个癌旁样本(一个病人可能对应多次生物学重复)。
8、(2)测序数据质量控制:对步骤(1)得到的测序原始数据进行质检。对于rna-seq数据:排除在所有样品中超过75%read计数小于1的低表达基因。对于甲基化数据:根据芯片检测的p值,针对某个cpg位点,若p>0.01的样本数超过总样本的50%,则将其过滤掉;此外,从分析中过滤掉含有snp的cpg位点、x和y染色体上的cpg以及发生交叉反应的探针上的cpg位点;为了最大限度地减少样品内和样品之间不必要的变异,对甲基化数据进行bmiq归一化;并将beta值转换为m值以方便后续差异甲基化位点(dmp)检测的执行。
9、(3)检测差异甲基化位点(dmp)和差异表达基因(deg):对步骤(2)得到的甲基化数据使用r软件包limma进行差异甲基化位点的检测分析,选择benjamin-hochberg方法调整p值,将p值<0.05,且同时甲基化水平均值差的绝对值>0.3的cpg位点作为候选的dmp。对于rna-seq数据,对原始的read计数进行log2(cpm(count)+1)标准化转换,使用r软件包limma进行差异表达基因的检测分析。将调整后的p值小于0.05和的基因作为差异表达的基因(deg)。
10、(4)挖掘结肠癌候选甲基化标志的cpg位点:首先用tcgabiolinks对illuminahuman450k甲基化芯片中的dmps进行注释分类;然后提取启动子区域附近(tss上游或者下游2kb p区域范围)的dmps和相应转录起始位点对应deg的表达数据进行联合分析。选择位于下调差异基因启动子中的高甲基化cpg位点(共1065个)作为进一步的候选cpg位点。通过lasso回归进行特征选择,最终选出最具有标志性的30个cpg位点。
11、(5)机器学习方法构建结肠癌诊断的数学模型:利用tcga中coad的数据样本中该30个位点的甲基化信息作为特征进行训练,利用支持向量机(svm)模型,采用10折交叉验证的方法确定模型的超参数,随后训练得到结肠癌诊断的数学模型。从geo公共数据库中下载三个独立的结肠癌的甲基化数据集。利用这三个独立的数据集对步骤(4)筛选出的结肠癌的甲基化标志性cpg位点进行外部验证,进一步确认本发明筛选出了可靠的结肠癌标志性的甲基化cpg位点。此外,候选的cpg位点所在的目标基因的表达数据被用来进行生存(预后)分析。
12、(6)methyltargt测序文库构建及测序:利用methyltargt测序方法对所选择的30个cpg位点在25对新的配对结肠癌组织数据中进行测序验证。
13、(7)测序数据分析:对步骤(6)得到的原始的测序数据进行质检。将经过质检的reads使用软件flash(flash:fast length adjustment of short reads to improvegenome assemblies)进行read1和read2的拼接。利用blast+工具比对到人类参考基因组上,筛选能够覆盖目标序列90%或有90%碱基能够完整比对到其目标序列的reads用于后续的分析。通过非监督聚类发现,以该30个位点作为标志测序的数据可以很好的将肿瘤和非肿瘤区分开。
14、本发明通过对tcga中295个病人的样本进行转录组和差异甲基化的整合分析,构建了结肠癌dna差异甲基化图谱,挖掘异常的甲基化位点,并进一步通过lasso回归的方法筛选出了结肠癌诊断的30个dna甲基化的标志位点。随后以这些位点作为标志物通过机器学习方法在geo的多个数据集中进行验证,均呈现出较高的精确度。最后利用methyltarget目标区域甲基化测序方法对该30个位点的甲基化进行特异性测序,其可以非常有效的区分25对结肠癌的样本(肿瘤和匹配的癌旁组织),这些标志物将有望成为结肠癌诊断筛查的重要手段。
15、本发明的有益效果是:
16、以本发明标记物为基础,构建结肠癌诊断的数学模型;该模型灵敏度高,特异性好,在三个独立的结肠癌geo公共数据库验证中auc均达到0.98,在25对结肠癌样本(肿瘤和匹配的癌旁组织)验证中auc高达1,诊断效果良好。综上所述,本发明公开的dna甲基化分子标志物具有良好的诊断指标特性,可以有效用于结肠癌的诊断,具有较高的临床应用和推广价值。
- 还没有人留言评论。精彩留言会获得点赞!