一种纳米药物载体甲氧基聚乙二醇-聚(L-谷氨酸钠)的制备方法与流程
一种纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法
技术领域
1.本技术涉及一种纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,属于抗肿瘤纳米药物载体技术领域。
背景技术:
2.纳米药物载体是一种粒径在10~1000nm的药物递送载体材料,通常由天然或合成高分子材料制成。药物可以溶解、分散或化学键合等方式载入其中,与传统药物制剂相比,具有较高的生物利用度、缓控释和肿瘤靶向性等特点;在提高药物治疗效果的同时,还可提高药物稳定性、减轻药物的不良反应;纳米药物载体是目前国际上生物医药领域的研究热点。
3.甲氧基聚乙二醇-聚(l-谷氨酸钠)(mpeg-p(l-glu-na))是一种线性嵌段共聚物,聚(l-谷氨酸钠)片段具有悬垂的-coona侧基,可用于药物结合和核心修饰,便于药物递送。文献(journal of controlled release 101(2005),223-232)报道mpeg-p(l-glu-na)可与顺铂、奥沙利铂形成具有抗肿瘤活性的配位化合物;本技术人之前的专利cn201910547554.6(一种顺铂微粒系统组成、制备方法及应用)提供了一种mpeg-p(l-glu-na)与顺铂形成的载药复合物,具有更高的载药量和更广泛的肿瘤分布效果,提高了顺铂的抗癌效果且毒性明显降低。日本nanocarrier公司开发的顺铂胶束制剂nc-6004正是用此类聚合物与顺铂发生配位反应制备的药物制剂,聚合物侧链上的羧酸阴离子(coo-)与顺铂配位形成疏水端,聚乙二醇链作为亲水端,形成复合物胶束溶液;nc-6004目前正在临床iii期,临床前数据表明其耐受性良好且在动物模型中具有与顺铂相似或优于顺铂的抗肿瘤效果,在动物体内能降低顺铂的肾毒性和神经毒性(british journal of cancer,2005,93,678-687)。
4.甲氧基聚乙二醇-聚(l-谷氨酸钠)(mpeg-p(l-glu-na))的化学结构式如下:目前有关mpeg-p(l-glu-na)的合成方法在文献(bioconjugate chem.1992,3,295-301; cancer research 63,8977-8983,2003; journal of controlled release 101 101(2005)223-232; j biomed.eng.,2006,23(4),786-789;nature nanotechnology, 2011,166 等)中进行了报道,合成方式是甲氧基聚乙二醇胺(mpeg-nh2)引发l-谷氨酸-γ-苄酯环内酸酐(blg-nca)室温下聚合得到甲氧基聚乙二醇-聚(l-谷氨酸-γ-苄酯)(mpeg-pblg),然后进行碱解或者酸解脱掉聚合物中pblg片段的γ位苄基得到甲氧基聚乙二醇-聚
(l-谷氨酸钠)(mpeg-p(l-glu-na)),反应式如下:上述合成路线中起始物料l-谷氨酸-γ-苄酯环内酸酐(blg-nca)可以由l-谷氨酸-γ-苄酯与三光气制备得到;另一个起始物料甲氧基聚乙二醇胺(mpeg-nh2)商品销售价格较高,并且目前还没有进行大规模生产的供应商,现有文献报道的mpeg-nh2的合成方法主要有如下四种:一、文献(makromol.chem., rapid commun.12,159-165(1991); journal of pharmaceuticals sciences 87(10),1998,1242-1248)以甲氧基聚乙二醇(mpeg-oh)为起始原料,先与对甲苯磺酰氯(tscl)反应得到甲氧基聚乙二醇对甲苯磺酸酯(mpeg-ots),再在高压釜中与氨水反应得到甲氧基聚乙二醇胺(mpeg-nh2),反应式如下:此路线的胺化反应需要在高压釜中进行,并且mpeg-ots会发生水解副反应得到mpeg-oh,导致产品纯度较低,有文献(中南药学 2014,12(6),549-551)报道在胺化反应中添加氯化铵来提高转化率,但收率不高,需要用柱层析硅胶纯化产品,不适合工业化大规模生产。
5.二、文献(j.biomater.sci.polymer edn.,14(12),1389-1400(2003);精细化工中间体,36(1),40-42,(2006); j biomed.eng. 2006,23(4),786-789)以甲氧基聚乙二醇(mpeg-oh)为起始原料,与对甲苯磺酰氯(tscl)反应得到甲氧基聚乙二醇对甲苯磺酸酯(mpeg-ots),再采用gabriel合成法与邻苯二甲酰亚胺钾反应、水合肼脱保护得到甲氧基聚乙二醇胺(mpeg-nh2),反应式如下:
och2ch2ch2nh-p(l-glu-na),其合成方法是以mpeg-oh为起始原料经丙氰化和还原胺化两步反应(us6875841)得到甲氧基聚乙二醇正丙胺(mpeg-och2ch2ch2nh2),再在无水溶剂中引发blg-nca进行聚合反应得到嵌段共聚物,最后碱水解脱掉苄基而制备得到mpeg-och2ch2ch2nh-p(l-glu-na)(wo2009157279),反应式如下:此路线合成步骤较多,raney ni的氢化还原反应需要在高压釜中进行,对反应设备要求高,总收率不高。
9.综上所述,目前文献报道的甲氧基聚乙二醇-聚(l-谷氨酸钠)(mpeg-p(l-glu-na))的合成路线较长,以mpeg-oh为起始原料,需要4-5步反应,生产成本较高;此外在聚合反应中,由于长链mpeg-nh2的碱性较弱,引发blg-nca进行聚合时间比较长,反应过程不易控制。因此开发一条低成本、低污染、易操作的适合工业化生产的工艺路线具有非常重要的意义。
技术实现要素:
10.为了解决上述问题,提供了一种全新的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)制备方法,该制备方法的路线起始原料廉价易得,污染少、操作简单,生产成本低,适合大规模工业化生产。
11.根据本技术的一个方面,提供了一种纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,合成路线如下:其中,n为20~500中的任意整数,m为10~60中的任意整数;包括以下步骤:(1)将2-氯乙胺盐酸盐在0-5℃下经过碱a处理后得到2-氯乙胺游离碱,再与l-谷
氨酸-γ-苄酯环内酸酐在第一溶剂中发生聚合反应得到式1产物;(2)将甲氧基聚乙二醇(mpeg-oh,ch3o(ch2ch2o)noh,n为20~500中的任意整数)在第二溶剂中经碱b作用后,与式1产物发生亲核取代反应得到式2产物;(3)将式2产物在第三溶剂中溶解,加入0.5-2mol/l的氢氧化钠水溶液碱解,残留水溶液真空干燥,得到甲氧基聚乙二醇-聚(l-谷氨酸钠)。
12.可选地,包括以下步骤:(1)将2-氯乙胺盐酸盐悬浮在乙醚中,降温到0-5℃,加入碱a搅拌反应0.5-1h,过滤,减压浓缩得到2-氯乙胺游离碱,再依次加入第一溶剂和l-谷氨酸-γ-苄酯环内酸酐,在氮气保护下室温反应24-26h,将反应液减压浓缩,倒入乙醚中,搅拌后抽滤,再用乙醚洗涤即得式1产物;(2)将甲氧基聚乙二醇溶解到第二溶剂中,降温到0-5℃,加入碱b的四氢呋喃溶液,直至反应液为绿色,且0.5h内不变色,再加入式1产物,搅拌0.5-1h,升至室温在氮气保护下搅拌反应24-26h,抽滤去除不溶物,滤液倒入乙醚中,再用无水乙醇重结晶,真空干燥后即得式2产物;(3)将式2产物在第三溶剂中溶解,室温下加入0.5-2mol/l的氢氧化钠水溶液搅拌反应24-26h,反应液减压浓缩除去挥发性溶剂,残留水溶液在0-5℃搅拌析出白色固体,过滤,45℃真空干燥得到甲氧基聚乙二醇-聚(l-谷氨酸钠)。
13.可选地,碱a为碳酸钠、碳酸钾、碳酸氢钠、三乙胺、二异丙基乙胺、吡啶中的任意一种。
14.优选地,碱a为三乙胺。
15.可选地,2-氯乙胺盐酸盐与碱a的摩尔比为1:(1.0~5.0)。优选地,2-氯乙胺盐酸盐与碱a的摩尔比为1:(1.5~2.5);最优选地,2-氯乙胺盐酸盐与碱a的摩尔比为1:2;可选地,2-氯乙胺盐酸盐与l-谷氨酸-γ-苄酯环内酸酐的摩尔比为1:(10~60),可以制备m=10-60任意一个式1产物clch2ch2nh-p(l-gluobn)m。
16.可选地,第一溶剂为四氢呋喃、二氯甲烷、n,n-二甲基甲酰胺、氯仿和二氧六环中的任意一种。优选地,第一溶剂为二氯甲烷或四氢呋喃。
17.可选地,l-谷氨酸-γ-苄酯环内酸酐与第一溶剂重量比为1:(2~50)。
18.优选地,l-谷氨酸-γ-苄酯环内酸酐与第一溶剂重量比为1:(5~20)。
19.优选地,碱b为萘钾,甲氧基聚乙二醇与碱b的摩尔比为1:1。
20.可选地,甲氧基聚乙二醇与式1产物的摩尔比为1:(0.9~1.1)。
21.优选地,甲氧基聚乙二醇与式1产物的摩尔比为1:(0.95-1.05)。
22.采用不同分子量的mpeg-oh做原料,可以制备m=10-60任意一个,n是20~500中任意一个的聚合物mpeg-pblg[ch3o(ch2ch2o)nch2ch2nh-p(l-gluobn)m(n=20~500,m=10~60)],最终可以制备m是10~60中任意一个,n是20~500中任意一个的聚合物mpeg-p(l-glu-na)[ch3o(ch2ch2o)nch2ch2nh-p(l-gluna)m(n=20~500,m=10~60)]。
[0023]
可选地,第二溶剂为四氢呋喃、二氧六环、乙二醇二甲醚中的任意一种;优选为乙二醇二甲醚;甲氧基聚乙二醇与第二溶剂的重量比为1:(2~22),优选为1:(5~15)。
[0024]
可选地,第三溶剂为四氢呋喃、甲醇和乙醇中的任意一种;式2产物与氢氧化钠水溶液的摩尔比为1:(1.2~2.0)。优选地,第三溶剂为四氢呋喃,式2产物与氢氧化钠水溶液的
摩尔比为1:(1.4~1.8)。
[0025]
本技术中,“室温”为25℃。
[0026]
本技术的有益效果包括但不限于:1.根据本技术的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,该方法使用2-氯乙胺盐酸盐、l-谷氨酸-γ-苄酯环内酸酐和甲氧基聚乙二醇作为起始原料,合成路线短,成本低、污染少,操作简单。
[0027]
2.根据本技术的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,通过使用2-氯乙胺盐酸盐,避免后续副反应的发生,通过限定2-氯乙胺盐酸盐与碱a的反应温度,使2-氯乙胺盐酸盐能够中和完全,2-氯乙胺的游离碱能稳定存在,并且其碱性比长链大分子的mpeg-nh2强,更容易引发blg-nca的聚合,反应时间短。
[0028]
3.根据本技术的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,通过采用萘钾作为mpeg-oh脱氢的强碱试剂,可通过反应液颜色变化确定反应终点,确保对pblg链没有降解破坏作用。
[0029]
4.根据本技术的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,通过限定2-氯乙胺盐酸盐与碱a的摩尔比,使得2-氯乙胺能够完全游离为自由碱;通过限定2-氯乙胺盐酸盐与l-谷氨酸-γ-苄酯环内酸酐的摩尔比,可以制备m=10-60任意一个的clch2ch2nh-p(l-gluobn)m。
[0030]
5.根据本技术的纳米药物载体甲氧基聚乙二醇-聚(l-谷氨酸钠)的制备方法,通过限定甲氧基聚乙二醇与式1产物的摩尔比,使得两个聚合物的偶联能够顺利进行;通过限定甲氧基聚乙二醇与第二溶剂的重量比,使得甲氧基聚乙二醇能完全溶解,并且保证碱化反应能平稳进行。
附图说明
[0031]
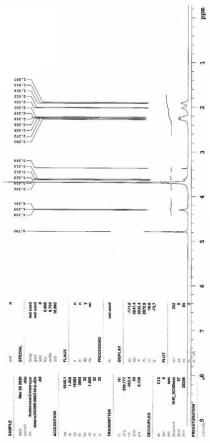
此处所说明的附图用来提供对本技术的进一步理解,构成本技术的一部分,本技术的示意性实施例及其说明用于解释本技术,并不构成对本技术的不当限定。在附图中:图1为本技术实施例1制备的mpeg
2000-p(l-glu-na)m(m=12-16)的1h nmr(d2o)图谱。
具体实施方式
[0032]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0033]
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。本发明所使用的试剂或原料均可通过常规途径购买获得,如无特殊说明,本发明所使用的试剂或原料均按照本领域常规方式使用或者按照产品说明书使用。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。本专利中所述的较佳实施方法与材料仅作示范之用。
[0034]
另外,高分子聚合物是以一定分子量范围为主要组成部分的混合物,本专利的mpeg-oh的数均分子量是2000,包含1800-2200的分子量范围。合成的聚谷氨酸片段也是一个范围,以下实施例合成实验中按照1:14投料进行反应,最后得到12-16个片段的谷氨酸片段数是正常范围。
[0035]
实施例1 甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)合成(mpeg
2000
表示mpeg-oh的mn为2000,ch3o(ch2ch2o)noh,n=42-48)制备方法包括以下步骤:(1)氯乙胺-聚(l-谷氨酸-γ-苄酯)(clch2ch2nh-pblg,clch2ch2nh-p(l-gluobn)m(m=12-16))的合成将2-氯乙胺盐酸盐(1.16g,0.01mol)悬浮在20ml乙醚中,降温到0℃,加入三乙胺(2.02g,0.02mol,2eq)搅拌反应0.5h,过滤,减压浓缩得到2-氯乙胺游离碱的淡黄色油状物;加入500ml二氯甲烷,缓慢加入l-谷氨酸-γ-苄酯环内酸酐(blg-nca,36.85g,0.14mol,14eq),氮气保护下室温搅拌反应24h;将反应液减压浓缩到1/4体积,倒入1l乙醚中,搅拌1h,抽滤,少许乙醚洗涤,得到27.6g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率87.6%。
[0036]
(2)甲氧基聚乙二醇-聚(l-谷氨酸-γ-苄酯)(mpeg-pblg,mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16))的合成将甲氧基聚乙二醇2000(mpeg
2000-oh,16g,0.008mol;mpeg-oh的mn为2000)溶解到400ml乙二醇二甲醚中,在0℃下加入适量萘钾(1.33g,0.008mol,1eq)的四氢呋喃溶液直至反应液显示绿色,在0.5h内不变色;然后慢慢加入clch2ch2nh-p(l-gluobn)m(m=12-16)(25.16g,0.008mol,1eq),继续保温搅拌1h,然后升到室温氮气保护搅拌反应24h;抽滤除去不溶物,滤液倒入4.0l乙醚中得到粗产品,然后用无水乙醇重结晶,真空干燥,得到30.5g白色固体产品mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16),收率74.6%;(3)甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)]的合成将mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16)(20g,苄酯摩尔数0.055mol)溶解到150ml四氢呋喃中,室温下加入0.5mol/l的氢氧化钠水溶液(165ml,0.0825mol,1.45eq),然后搅拌反应24h;反应液减压浓缩除去挥发性溶剂四氢呋喃,残留水溶液在0℃搅拌析出白色固体,过滤,45℃真空干燥,得到13.4g白色固体产品mpeg
2000-p(l-glu-na)m(m=12-16),收率82.4%。
[0037]
图1为目标产品核磁图谱,1h nmr(d2o, ppm): 1.8-2.4(br,p(l-glu-na)-ch2ch
2-);3.3(s,peg-ch3);3.5-3.8(br,peg-ch2ch
2-o-); 4.2-4.4(br,p(l-glu-na)-ch-)。从图1中可以看出,所得产品的结构是mpeg与pglu的共聚物。
[0038]
实施例2 甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)]的合成(mpeg
2000
表示mpeg-oh的mn为2000,ch3o(ch2ch2o)noh,n=42-48)制备方法包括以下步骤:(1)氯乙胺-聚(l-谷氨酸-γ-苄酯)(clch2ch2nh-pblg,clch2ch2nh-p(l-gluobn)m(m=12-16))的合成将2-氯乙胺盐酸盐(2.32g,0.02mol)悬浮在20ml乙醚中,降温到2℃,加入三乙胺(3.03g,0.03mol)搅拌反应0.5h,过滤,减压浓缩得到2-氯乙胺游离碱的淡黄色油状物;加入1500ml二氯甲烷,缓慢加入l-谷氨酸-γ-苄酯环内酸酐(blg-nca,73.7g,0.28mol,14eq),氮气保护下室温搅拌反应24h;将反应液减压浓缩到1/4体积,倒入2l乙醚中,搅拌1h,抽滤,少许乙醚洗涤,得到51.9g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率81.5%。
[0039]
(2)甲氧基聚乙二醇-聚(l-谷氨酸-γ-苄酯)(mpeg-pblg,mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16))的合成将甲氧基聚乙二醇2000(mpeg
2000-oh,16g,0.008mol,1eq;mpeg-oh的mn为2000)溶解到400ml乙二醇二甲醚中,在2℃下加入适量萘钾(约1.33g,0.008mol,1eq)的四氢呋喃溶液直至反应液显示绿色,在0.5h内不变色;然后慢慢加入clch2ch2nh-p(l-gluobn)m(m=12-16)(22.64g,0.0072mol,0.9eq),继续保温搅拌1h,然后升到室温氮气保护搅拌反应24h;抽滤除去不溶物,滤液倒入4.0l乙醚中得到粗产品,然后用无水乙醇重结晶,真空干燥,得到27.2g白色固体产品mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16),收率66.6%;(3)甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)]的合成将mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16)(20g,苄酯摩尔数0.055mol)溶解到150ml甲醇中,室温下加入1.0mol/l的氢氧化钠水溶液(110ml,0.11mol,2eq),然后搅拌反应24h;反应液减压浓缩除去挥发性溶剂四氢呋喃,残留水溶液在2℃搅拌析出白色固体,过滤,45℃真空干燥,得到11.2g白色固体产品mpeg
2000-p(l-glu-na)m(m=12-16),收率68.9%。
[0040]
实施例3 甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)]的合成(mpeg
2000
表示mpeg-oh的mn为2000,ch3o(ch2ch2o)noh,n=42-48)制备方法包括以下步骤:(1)氯乙胺-聚(l-谷氨酸-γ-苄酯)(clch2ch2nh-pblg,clch2ch2nh-p(l-gluobn)m(m=12-16))的合成将2-氯乙胺盐酸盐(2.32g,0.02mol)悬浮在20ml乙醚中,降温到5℃,加入三乙胺(5.05g,0.05mol,2.5eq)搅拌反应0.5h,过滤,减压浓缩得到2-氯乙胺游离碱的淡黄色油状物;加入1000ml四氢呋喃,缓慢加入l-谷氨酸-γ-苄酯环内酸酐(blg-nca,73.7g,0.28mol,14eq),氮气保护下室温搅拌反应24h;将反应液减压浓缩到1/4体积,倒入2l乙醚中,搅拌1h,抽滤,少许乙醚洗涤,得到50.9g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率79.9%。
[0041]
(2)甲氧基聚乙二醇-聚(l-谷氨酸-γ-苄酯)(mpeg-pblg,mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16))的合成将甲氧基聚乙二醇2000(mpeg
2000-oh,16g,0.008mol,1eq;mpeg-oh的mn为2000)溶解到400ml乙二醇二甲醚中,在5℃下加入适量萘钾(约1.33g,0.008mol,1eq)的四氢呋喃溶液直至反应液显示绿色,在0.5h内不变色;然后慢慢加入clch2ch2nh-p(l-gluobn)m(m=12-16)(27.68g,0.0088mol,1.1eq),继续保温搅拌1h,然后升到室温氮气保护搅拌反应24h;抽滤除去不溶物,滤液倒入4.0l乙醚中得到粗产品,然后用无水乙醇重结晶,真空干燥,得到28.6g白色固体产品mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16),收率70.0%;(3)甲氧基聚乙二醇-聚(l-谷氨酸钠)[mpeg
2000-p(l-glu-na)m(m=12-16)]的合成将mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16)(20g,苄酯摩尔数0.055mol)溶解到150ml乙醇中,室温下加入2.0mol/l的氢氧化钠水溶液(33ml,0.66mol,1.2eq),然后搅拌反应24h;反应液减压浓缩除去挥发性溶剂四氢呋喃,残留水溶液在5℃搅拌析出白色固体,过滤,45℃真空干燥,得到10.7g白色固体产品mpeg
2000-p(l-glu-na)m(m=12-16),收率65.8%。
[0042]
对比例1对比例1与实施例1的区别在于:在步骤(1)中,对比例1中使用2-溴乙胺盐酸盐代替2-氯乙胺盐酸盐。
[0043]
结果为步骤(1)最终得到13.8g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率43.8%。
[0044]
对比例2对比例2与实施例1的区别在于:在步骤(1)中,对比例2的2-氯乙胺盐酸盐与碱a的反应温度为20℃。
[0045]
结果为步骤(1)最终得到17.4g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率55.2%。
[0046]
对比例3对比例3与实施例1的区别在于:在步骤(1)中,对比例3的2-氯乙胺盐酸盐与碱a的反应温度为-5℃。
[0047]
结果为步骤(1)最终得到18.2g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率57.8%。
[0048]
对比例4对比例4与实施例1的区别在于:对比例4中碱b为氢氧化钾。
[0049]
结果为步骤(2)中甲氧基聚乙二醇2000与clch2ch2nh-p(l-gluobn)m(m=12-16)不反应,无法得到产品。
[0050]
对比例5对比例5与实施例1的区别在于:在步骤(2)中,对比例5中甲氧基聚乙二醇与式1产物的摩尔比为1:1.3。
[0051]
最终步骤(2)得到25.9g白色固体产品mpeg
2000-och2ch2nh-p(l-gluobn)m(m=12-16),收率63.4%。
[0052]
对比例6对比例6与实施例1的区别在于:在步骤(1)中,对比例6中2-氯乙胺盐酸盐与碱a的摩尔比为1:0.8。
[0053]
结果为步骤(1)最终得到21.8g白色固体产品clch2ch2nh-p(l-gluobn)m(m=12-16),收率69.4%。
[0054]
由上述实施例和对比例的结果表明,经过本技术所限定的方法所制备的甲氧基聚乙二醇-聚(l-谷氨酸钠)路线起始原料廉价易得,污染少、操作简单,生产成本低,适合大规模工业化生产。
[0055]
对比例1中使用2-溴乙胺盐酸盐代替2-氯乙胺盐酸盐,最终结果表明其收率低,分析原因为在步骤(2)中部分溴会被碱取代变为羟基,引起了副反应的发生,产品收率偏低;对比例2中2-氯乙胺盐酸盐与碱a的反应温度高于本技术限定范围,产品收率低,分析原因为温度高2-氯乙胺会与碱(三乙胺)反应变为季铵盐;对比例3中2-氯乙胺盐酸盐与碱a的反应温度低于本技术限定范围,产品收率低,分析原因为温度较低使2-氯乙胺盐酸盐中和不完全;对比例4中碱b为氢氧化钾,甲氧基聚乙二醇无法与式1产物进行反应,原因为氢氧化钾的碱性弱,无法与甲氧基聚乙二醇的端位羟基进行反应。
[0056]
对比例5中使用的为甲氧基聚乙二醇与式1产物的摩尔比不在本技术限定范围内,产品收率低,分析原因为式1产物剩余较多,无法有效参与反应;对比例6中2-氯乙胺盐酸盐与碱a的摩尔比不在本技术限定范围内,产品收率低,分析原因为2-氯乙胺不能完全游离为自由碱。
[0057]
以上所述,仅为本技术的实施例而已,本技术的保护范围并不受这些具体实施例的限制,而是由本技术的权利要求书来确定。对于本领域技术人员来说,本技术可以有各种更改和变化。凡在本技术的技术思想和原理之内所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1