一种邻-碳硼烷取代的氧杂环丁烷及其衍生物的制备方法
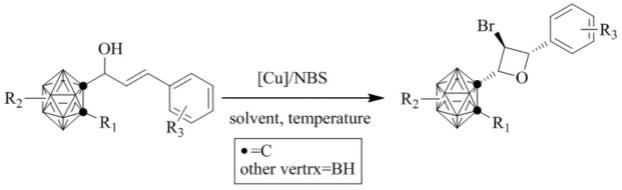
1.本发明属于有机合成化学技术领域,涉及一种邻-碳硼烷取代的氧杂环丁烷及其衍生物的制备方法。
背景技术:
2.邻-碳硼烷是一种由两个碳原子、十个硼原子和十二个氢原子组成的具有三维芳香性的正二十面体分子簇合物。因其特殊的几何构形、成键方式、超高的硼含量以及高度缺电子等性质,邻-碳硼烷及其衍生物被广泛应用于硼中子俘获治疗(bnct)、药物开发、超分子自组装、配位化学、催化、储能、有机光电材料等领域。因此,开展邻-碳硼烷衍生物的合成方法学研究对推进碳硼烷化学的发展及其在相关领域的应用具有重要意义。
3.氧杂环丁烷是一类含有四元环醚结构的化合物,不仅是重要的有机合成中间体,也是天然和合成的具有抗癌、抗hiv、抑制谷氨酰胺合成酶等多种生物或药物活性分子结构中的重要活性单元。由于氧杂环丁烷属于含能杂环化合物,因而在含能粘合剂、火药、生物农药等领域也具有较多的应用。因此,开展氧杂环丁烷衍生物的多样化合成具有重要的学术意义和应用价值。
4.氧杂环丁烷作为含氧的饱和四元杂环,氧原子上的孤对电子使其可以作为路易斯碱和氢键受体。基于这个特点,氧杂环丁烷常作为偕二甲基和羰基的生物电子等排体,用于改善药物分子的物理化学和药代动力学性质。现有技术利用肉桂醇制备芳基氧杂环丁烷主要采用双(2,4,6-三甲基吡啶)溴代六氟磷酸盐作为溴源形成鎓离子,从而诱导分子内开环重排形成氧杂环丁烷,此种溴盐不仅价格昂贵而且储存条件极为苛刻,不具备工业放大的潜力和优势。
5.研究结果表明,将药物分子中的芳香环用具有三维芳香性的邻-碳硼烷代替后,不仅可以提高药物分子的生物活性,还能够发现诸多未知的新的特点;且以此合成的新化合物可能在医药、材料等领域具有全新的应用价值。然而,目前尚未见有关邻-碳硼烷取代的氧杂环丁烷的合成方法。因此,开展邻-碳硼烷取代的氧杂环丁烷的多样化合成具有重要意义。
技术实现要素:
6.针对邻-碳硼烷取代的氧杂环丁烷在医药和材料等领域的重要应用前景,本发明的目的在于提供一种操作简单、合成条件温和、适用范围广、低成本的邻-碳硼烷取代的氧杂环丁烷及其衍生物的制备方法。
7.为了实现本发明的这些目的,提供了一种邻-碳硼烷取代的氧杂环丁烷衍生物,其特征在于,结构通式为:
[0008][0009]
其中,取代基r1为氢原子、甲基、苯基以及4-甲基苯基中的任意一种;r2为取代苯基或氢原子;r3为卤素、硝基、氢原子中的任意一种。
[0010]
本发明还提供一种如上所述的邻-碳硼烷取代的氧杂环丁烷衍生物的制备方法,包括:
[0011]
以为原料,在催化剂、有机溶剂以及n-溴代丁二酰亚胺存在下进行反应,实现分子内串联环化得到邻-碳硼烷取代的氧杂环丁烷衍生物,即
[0012]
取代基r1为氢原子、甲基、苯基以及4-甲基苯基中的任意一种;r2为取代苯基或氢原子;r3为卤素、硝基、氢原子中的任意一种。
[0013]
优选的是,所述催化剂为三氟甲磺酸铜;所述有机溶剂为1,2-二氯乙烷、四氢呋喃或乙腈中的任意一种;所述反应的温度为80~90℃;所述反应的气氛为氩气。
[0014]
优选的是,在干燥的反应容器中,氩气保护下加入原料有机溶剂,催化剂以及n-溴代丁二酰亚胺,将反应容器置于80~90℃中反应,用薄层色谱监测反应的进行,反应结束后将体系过滤,滤液经减压蒸馏除去多余溶剂,经过柱层析分离得到目标产物
[0015]
优选的是,所述与有机溶剂的摩尔体积比为1mmol:8~
12ml;所述与催化剂的摩尔比为1:0.3~0.6;所述与n-溴代丁二酰亚胺的摩尔比为1:1~3;所述过滤采用短硅胶柱过滤,乙酸乙酯作为淋洗剂;柱层析分离采用的洗脱剂为体积比为15~25:1的石油醚和乙酸乙酯。
[0016]
本发明以廉价的n-溴代丁二酰亚胺(nbs)作为溴源,在铜盐的催化下可实现邻-碳硼烷取代的氧杂环丁烷衍生物的高效构建,为探索其在医药、材料等领域的应用提供物质和技术基础。
[0017]
本发明的邻-碳硼烷取代的氧杂环丁烷及其衍生物的制备方法的反应式为:
[0018][0019]
其中,取代基r1为氢原子、甲基、苯基以及4-甲基苯基中的任意一种;r2为取代苯基或氢原子;r3为卤素、硝基、氢原子中的任意一种。
[0020]
本发明至少包括以下有益效果:
[0021]
(1)本发明设计的邻-碳硼烷取代的氧杂环丁烷衍生物的制备方法具有原料廉价易得、合成条件简单、反应产率高、反应普适性好等特点;
[0022]
(2)本发明的邻-碳硼烷取代的氧杂环丁烷衍生物制备方法为探索其在医药、材料等领域的应用提供了物质和技术基础。
[0023]
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
附图说明:
[0024]
图1为本发明实施例1中3a的1hnmr;
[0025]
图2为本发明实施例1中3a的
13
cnmr;
[0026]
图3为本发明实施例2中3b的1hnmr
[0027]
图4为本发明实施例2中3b的
13
cnmr
[0028]
图5为本发明实施例4中3d的1hnmr
[0029]
图6为本发明实施例4中3d的
13
cnmr
[0030]
图7为本发明实施例6中3f的1hnmr;
[0031]
图8为本发明实施例6中3f的
13
cnmr;
[0032]
图9为本发明实施例7中3g的1hnmr;
[0033]
图10为本发明实施例7中3g的
13
cnmr;
[0034]
图11为本发明实施例8中3h的1hnmr;
[0035]
图12为本发明实施例8中3h的
13
cnmr;
[0036]
图13为本发明实施例9中3i的1hnmr;
[0037]
图14为本发明实施例9中3i的
13
cnmr。
具体实施方式:
[0038]
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
[0039]
应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不配出一个或多个其它元件或其组合的存在或添加。
[0040]
本发明的邻-碳硼烷取代的氧杂环丁烷及其衍生物的制备方法的反应式为:
[0041][0042]
其中,取代基r1为氢原子、甲基、苯基以及4-甲基苯基中的任意一种;r2为取代苯基或氢原子;r3为卤素、硝基、氢原子中的任意一种。
[0043]
实施例1:
[0044]
制备原料1-肉桂醇-邻-碳硼烷:
[0045][0046]
氩气氛围下,在配备有磁力搅拌子和冷凝管的干燥250ml圆底烧瓶中,依次加入邻-碳硼烷720mg(5mmol),50ml干燥的四氢呋喃;将反应瓶浸入-78℃的冷凝浴中并开始搅拌,待体系温度降至-78℃以后,使用注射器缓慢加入正丁基锂3.1ml(1.6m,5.5mmol);体系在-78℃下反应30分钟后加入肉桂醛859mg(5.5mmol),并在-78℃下继续反应3小时,使用tlc板监测反应进度,反应结束后,加水淬灭反应,随后使用乙酸乙酯萃取反应,依次使用饱和氯化铵水溶液,饱和食盐水洗涤,收集有机相,加无水硫酸钠干燥,干燥一小时后,浓缩有机相,采用柱层析技术分离产物,洗脱剂为石油醚:乙酸乙酯=10:1,得到目标产物825mg,产率55%。
[0047]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.41-7.31(m,5h),6.65-6.62(d,1h,j=15hz),6.11-6.06(dd,1h,j=5hz,15hz),4.77-4.75(m,1h),4.08(s,1h),2.29(s,1h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ135.0,134.8,128.9,128.8,126.9,126.2,77.7,73.7,58.4;
11
b{1h}nmr(160mhz,cdcl3,ppm):-3.2(2b),-4.3(1b),-8.8(1b),-9.1(1b),-12.3(2b),-13.1(1b),-13.8(1b),-14.5(1b).
[0048]
制备邻-碳硼烷取代的氧杂环丁烷的条件优化:
[0049]
以1-肉桂醇-邻-碳硼烷(2a)为原料制备邻-碳硼烷取代的氧杂环丁烷(3a)为例,对反应条件进行优化如下表1,其中dce为1,2-二氯乙烷,thf为四氢呋喃:
[0050][0051]
表1
[0052][0053]
经过条件筛选后,筛选出产物3a的最优条件为编号1:在氩气气氛下反应体系内加入50mol%三氟甲磺酸铜,1倍当量n-溴代丁二酰亚胺,在1,2-二氯乙烷中80℃反应6.5小时。典型操作步骤如下:
[0054]
3a的制备
[0055][0056]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-肉桂醇-邻-碳硼烷(27.6mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(nbs,17.8mg,0.1mmol)、三氟甲磺酸铜(cu(otf)2,18.1mg,0.05mmol),将体系容器置于80℃油浴中反应,tlc薄层板检测反应的进行,反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物23.6mg,产率67%。
[0057]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.44-7.41(m,3h),7.38-7.36(m,2h),5.63-5.62(d,1h,j=5hz),5.05-5.04(d,1h,j=5hz),4.49-4.46(dd,1h,j=5hz),3.90(s,1h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ136.9,129.6,129.0,125.5,86.7,83.8,73.8,58.7,
(m-h)-387.11600,found 387.11673.
[0068]
实施例4:
[0069]
3d的制备:
[0070][0071]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-(4-溴)-肉桂醇-邻-碳硼烷(35.4mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物30.2mg,产率70%。
[0072]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.57-7.56(d,2h,j=5hz),7.25-7.23(d,2h,j=10hz),5.59-5.58(d,1h,j=5hz),5.04-5.02(d,1h,j=10hz),4.43-4.40(dd,1h,j=10hz,5hz),3.86(s,1h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ136.0,132.2,126.9,123.7,86.0,83.9,58.7,45.7;
11
b{1h}nmr(160mhz,cdcl3,ppm):δ-2.5(2b),-3.4(2b),-8.7(2b),-12.3(1b),-13.1(2b),-14.1(1b);hrms(esi)m/z calculated for c
11h17b10
br2o-(m-h)-433.06344,found 433.06342.
[0073]
实施例5:
[0074]
3e的制备:
[0075][0076]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-(4-硝基)-肉桂醇-邻-碳硼烷(32.1mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mmol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物18.8mg,产率47%。
[0077]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ8.31-8.30(d,2h,j=10hz),7.55-7.53(d,2h,j=10hz),5.77-5.76(d,1h,j=5hz),5.11-5.09(d,1h,j=10hz),4.44-4.41(dd,1h j=10hz,5hz),3.85(s,1h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ143.9,139.8,125.7,124.3,85.1,84.4,65.1,58.7,45.3;
11
b{1h}nmr(192mhz,cdcl3,ppm):δ-2.5(2b),-3.3(2b),-8.7(2b),-12.4(1b),-13.1(2b),-14.1(1b).hrms(esi)m/z calculated for c
11b10h17
no3br-(m-h)-398.14005,found 398.14066.
[0078]
实施例6:
[0079]
制备1-肉桂醇-2-甲基-邻-碳硼烷:
[0080][0081]
氩气氛围下,在配备有磁力搅拌子和冷凝管的干燥250ml圆底烧瓶中,依次加入1-甲基-邻-碳硼烷790mg(5mmol),50ml干燥的四氢呋喃。将反应瓶浸入-78℃的冷凝浴中并开始搅拌,待体系温度降至-78℃以后,使用注射器缓慢加入正丁基锂3.1ml(1.6m,5.5mmol)。体系在-78℃下反应30分钟后加入肉桂醛859mg(5.5mmol),并在-78℃下继续反应3小时,使用tlc板监测反应进度。反应结束后,加水淬灭反应。随后使用乙酸乙酯萃取反应,依次使用饱和氯化铵水溶液,饱和食盐水洗涤。收集有机相,加无水硫酸钠干燥。干燥一小时后,浓缩有机相,采用柱层析技术分离产物,洗脱剂为石油醚:乙酸乙酯=10:1,得到目标产物1-肉桂醇-2-甲基-邻-碳硼烷。
[0082]
3f的制备:
[0083][0084]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-肉桂醇-2-甲基-邻-碳硼烷(29.0mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mmol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物22.1mg,产率60%。
[0085]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.46-7.40(m,5h),5.65-5.64(d,1h,j=5hz),5.07-5.05(d,1h,j=5hz),4.58-4.55(dd,1h,j=10hz,5hz),2.09(s,3h);
13
c{1h}nmr(100mhz,cdcl3,ppm):δ137.2,129.6,128.9,125.8,87.2,82.3,73.7,46.8,23.1;
11
b{1h}nmr(160mhz,cdcl3,ppm):δ-2.0(2b),-5.1(2b),-9.5(2b),-10.2(2b),-12.0(2b).
[0086]
实施例7:
[0087]
制备1-肉桂醇-2-苯基-邻-碳硼烷:
[0088][0089]
氩气氛围下,在配备有磁力搅拌子和冷凝管的干燥250ml圆底烧瓶中,依次加入1-苯基-邻-碳硼烷1100mg(5mmol),50ml干燥的四氢呋喃。将反应瓶浸入-78℃的冷凝浴中并开始搅拌,待体系温度降至-78℃以后,使用注射器缓慢加入正丁基锂3.1ml(1.6m,5.5mmol)。体系在-78℃下反应30分钟后加入肉桂醛859mg(5.5mmol),并在-78℃下继续反应3小时,使用tlc板监测反应进度。反应结束后,加水淬灭反应。随后使用乙酸乙酯萃取反
应,依次使用饱和氯化铵水溶液,饱和食盐水洗涤。收集有机相,加无水硫酸钠干燥。干燥一小时后,浓缩有机相,采用柱层析技术分离产物,洗脱剂为石油醚:乙酸乙酯=10:1,得到目标产物1-肉桂醇-2-苯基-邻-碳硼烷。
[0090]
3g的制备:
[0091][0092]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-肉桂醇-2-苯基-邻-碳硼烷(35.2mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mmol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物17.2mg,产率40%。
[0093]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.71-7.69(d,2h,j=10hz),7.53-7.49(dd,1h,j=10hz,10hz),7.44-7.41(m,7h),5.41-5.39(d,1h,j=10hz),4.53-4.49(dd,1h,j=10hz,10hz),4.40-4.39(d,1h,j=5hz);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ137.3,131.2,131.1,129.6,129.5,129.0,128.8,125.8,86.6,82.6,81.4,80.8,46.7;
11
b{1h}nmr(160mhz,cdcl3,ppm):δ-1.3(2b),-3.3(2b),-9.6(2b),-10.6(2b),-12.2(2b).
[0094]
实施例8:
[0095]
制备1-肉桂醇-2-(4-甲基)-苯基-邻-碳硼烷:
[0096][0097]
氩气氛围下,在配备有磁力搅拌子和冷凝管的干燥250ml圆底烧瓶中,依次加入1-(4-甲基)-苯基-邻-碳硼烷1170mg(5mmol),50ml干燥的四氢呋喃。将反应瓶浸入-78℃的冷凝浴中并开始搅拌,待体系温度降至-78℃以后,使用注射器缓慢加入正丁基锂3.1ml(1.6m,5.5mmol)。体系在-78℃下反应30分钟后加入肉桂醛859mg(5.5mmol),并在-78℃下继续反应3小时,使用tlc板监测反应进度。反应结束后,加水淬灭反应。随后使用乙酸乙酯萃取反应,依次使用饱和氯化铵水溶液,饱和食盐水洗涤。收集有机相,加无水硫酸钠干燥。干燥一小时后,浓缩有机相,采用柱层析技术分离产物,洗脱剂为石油醚:乙酸乙酯=10:1,得到目标产物1-肉桂醇-2-(4-甲基)-苯基-邻-碳硼烷。
[0098]
3h的制备:
[0099][0100]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-肉桂醇2-(4-甲基)-苯基-邻-碳硼烷(36.6mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mmol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物24.9mg,产率56%。
[0101]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.57-7.55(d,2h,j=10hz),7.41-7.40(m,5h),7.22-7.20(d,2h,j=10hz),5.41-5.39(d,1h,j=10hz),4.52-4.49(dd,1h,j=5hz,10hz),4.41-4.40(d,1h,j=5hz),2.40(s,3h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ141.6,137.3,131.1,129.7,129.5,128.8,126.8,125.8,86.5,82.8,81.4,80.8,46.7,21.1;
11
b{1h}nmr(160mhz,cdcl3,ppm):δ-1.3(2b),-3.5(2b),-9.7(2b),-10.7(2b),-12.3(2b).
[0102]
实施例9:
[0103]
制备1-肉桂醇-9,12-二-(4-氯-苯基)-邻-碳硼烷:
[0104][0105]
氩气氛围下,在配备有磁力搅拌子和冷凝管的干燥250ml圆底烧瓶中,依次加入9,12-二-(4-氯-苯基)-邻-碳硼烷1825mg(5mmol),50ml干燥的四氢呋喃。将反应瓶浸入-78℃的冷凝浴中并开始搅拌,待体系温度降至-78℃以后,使用注射器缓慢加入正丁基锂3.1ml(1.6m,5.5mmol)。体系在-78℃下反应30分钟后加入肉桂醛859mg(5.5mmol),并在-78℃下继续反应3小时,使用tlc板监测反应进度。反应结束后,加水淬灭反应。随后使用乙酸乙酯萃取反应,依次使用饱和氯化铵水溶液,饱和食盐水洗涤。收集有机相,加无水硫酸钠干燥。干燥一小时后,浓缩有机相,采用柱层析技术分离产物,洗脱剂为石油醚:乙酸乙酯=10:1,得到目标产物1-肉桂醇-9,12-二-(4-氯-苯基)-邻-碳硼烷。
[0106]
3i的制备:
[0107][0108]
在氩气保护下,向干燥的10ml圆底烧瓶中依次加入1-肉桂醇-9,12-二-(4-氯-苯基)-邻-碳硼烷(49.7mg,0.1mmol)、1,2-二氯乙烷(1ml)、n-溴代丁二酰亚胺(17.8mg,
0.1mmol)、三氟甲磺酸铜(18.1mg,0.05mmol),将反应容器置于80℃油浴中反应,tlc薄层板检测反应的进行。反应结束后,短硅胶柱过滤,乙酸乙酯作为淋洗剂,将滤液减压浓缩后,柱色谱分离提纯,用石油醚/乙酸乙酯=20:1做洗脱剂,柱色谱分离得到产物23.0mg,产率40%。
[0109]
核磁数据:1h nmr(500mhz,cdcl3,ppm):δ7.44-7.42(m,3h),7.38-7.37(m,2h),7.10-7.09(m,8h),5.67-5.66(d,1h,j=5hz),5.17-5.16(d,1h,j=5hz),4.52-4.49(dd,1h,j=5hz,10hz),4.01(s,1h);
13
c{1h}nmr(125mhz,cdcl3,ppm):δ136.8,134.2,134.1,133.7,133.6,129.7,129.0,127.5,125.4,86.8,83.6,69.4,53.9,46.0;
11
b{1h}nmr(160mhz,cdcl3,ppm):δ7.2(2b),6.4(2b),-8.9(2b),-13.4(4b);hrms:calculated for c
23b10h24
obrcl
2-(m-h)-575.13758,found 575.13867.
[0110]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1