一种微生物物种高通量鉴定解析方法
1.本发明涉及微生物菌种鉴定领域,具体涉及一种微生物物种高通量鉴定解析方法。
背景技术:
2.随着微生物高通量测序技术成熟,通过通量测定微生物16s rrna基因序列鉴定分离获得的微生物种属信息已成为现实。目前,通常做法是将多个微生物16s rrna序列进行混合测序,从而降低成本。为了能从混合样本中准确识别对应微生物的16s rrna信息,常在引物序列两端添加特异性标签标记微生物16s rrna序列。然而,无论测序成本如何节约,必须给每一个分离得到的微生物分配一个特异性序列标签,且需要对每一个微生物16s rrna进行单独扩增。繁杂的扩增过程及特异性引物定制都将耗费大量的人力和物力成本。
技术实现要素:
3.本发明的目的是提供一种可准确解析每个微生物对应的种属信息,极大降低了微生物物种鉴定过程中的人力和物力成本的微生物物种高通量鉴定解析方法。
4.本发明的微生物物种高通量鉴定解析方法是通过向混合样品中添加特异性16s rrna人工序列,结合倍数混样,能够做到仅需对多个微生物混合样品进行2次混合测序,即可准确解析每个微生物对应的种属信息,极大降低了微生物物种鉴定过程中的人力和物力成本,从而实现了本发明的目的。
5.本发明微生物物种高通量鉴定解析方法,将各待测样本混合形成混合样,混合样至少有2个,不同混合样中各待测样本浓度设置倍数差,且每个样本的倍数差均不同,同时向混合样品中加入特异性16s rrna人工序列,采用引物515f:5
’‑
gtgccagcmgccgcggtaa-3’和806r:5
’‑
ggactachvgggtwtctaat-3’对混合样中16s rrna进行pcr扩增,反应产物进行测序拼接,基于数据库对微生物物种进行注释,获得待测样品的微生物的分类信息;
6.所述的特异性16s rrna人工序列是5
’‑
gtgtcagcagccgcggtaatacgaagggggc tagcgttgtacatgcctgcgtgtcgtaaagggcgcgcaggcggctttgtaagttgggcgtgaaaggcctgggcttaacccgggaatggctgtgtgtttggtaaggctcgagtacgggagaggatggtggaattcccagtgtagaggtgaaattcgtagatattaggtggttgattgcgtgtcaaggcggccatctggaccgtaactgacgctgaggcgcgaaagcgtggggagcaaacaggattagaaacccgcgtagtcc-3’。
7.优选,所述的pcr扩增,其反应体系为12.5μl taq酶反应液,0.5μl上下游引物,2μl模板dna和9.5μl h2o。
8.优选,所述的pcr扩增,其反应条件为95℃ 5min,28个循环(95℃ 30s,56℃ 30s,72℃ 30s),72℃ 5min。
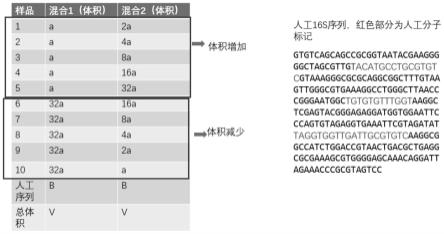
9.优选,两个混合样中,混合样2与混合样1相比,一半样品体积增加,一半样品体积减少,特异性16s rrna人工序列的体积和总体积恒定;任意一样品在混合样2与混合样1中体积比值与其他样品均不同,且差异至少1.5倍以上。
10.优选,所述的反应产物进行测序是反应产物采用hiseq 2500pe250平台测序。
11.优选,所述的拼接是基于vsearch平台进行拼接,并基于qiime2平台,采用dada2算法进行质控。
12.优选,所述的基于数据库对微生物物种进行注释是基于silva数据库对微生物物种进行注释,获得微生物的分类信息。
13.本发明相对于现有技术具有如下的优点及效果
14.1.仅需对多个微生物混合样品进行2次混合测序,即可准确解析每个微生物对应的种属信息,克服了大量微生物物种鉴定时存在的高成本问题,极大地降低了人力、物力成本。
15.2.目前测序结果准确性评估依然存在问题,添加的人工序列可作为内标,用于纠正测序过程中产生的错误。使用添加的人工序列优化数据分析参数,提高鉴定的准确性。
附图说明
16.图1是样品添加示意图,是以10个样品混合为例,混合2与混合1相比,一半样品体积增加,一半样品体积减少,人工序列体积和总体积恒定;任意一样品在混合2与混合1中体积比值与其他样品均不同,且差异至少1.5倍以上。
具体实施方式
17.以下实施例是对本发明的进一步说明,而不是对本发明的限制。
18.实施例1:
19.1、测试微生物:随机选用前期实验室分选待鉴定的微生物10株接种至lb固体培养基平皿上,37℃培养10h,获得10个单菌落。
20.2、微生物悬液制备:挑取平皿上培养获得的微生物单菌落至10个pcr管中,加入100μl超纯水,加盖后100℃裂解10min,获得微生物悬液。
21.3、混合样本制备:按照表1吸取微生物悬液,获得混合样本1和混合样本2。
22.表1:混合样本制备体积参照表
23.[0024][0025]
(3)人工16s rrna序列添加:分别向混合样本1和混合样本2中添加100ng/μl人工16srrna序列10μl,获得用于扩增的混合样本1和2。
[0026]
16s rrna人工序列:5
’‑
gtgtcagcagccgcggtaatacgaagggggctagcgttgtacatgcctgcgtgtcgtaaagggcgcgcaggcggctttgtaagttgggcgtgaaaggcctgggcttaacccgggaatggctgtgtgtttggtaaggctcgagtacgggagaggatggtggaattcccagtgtagaggtgaaattcgtagatattaggtggttgattgcgtgtcaaggcggccatctggaccgtaactgacgctgaggcgcgaaagcgtggggagcaaacaggattagaaacccgcgtagtcc-3’。
[0027]
(4)16s rrna扩增:采用引物515f(5
’‑
gtgccagcmgccgcggtaa-3’)和806r(5
’‑
ggactachvgggtwtctaat-3’)对混合样本中16s rrna进行扩增。反应体系为12.5μltaq酶反应液,0.5μl引物,2μl模板dna(待扩增混合样本1、2)和9.5μl h2o。反应条件为95℃ 5min,28个循环(95℃ 30s,56℃ 30s,72℃ 30s),72℃ 5min。反应产物采用hiseq2500pe250平台测序。
[0028]
(5)数据分析及物种解析:下机数据基于vsearch平台进行拼接,并基于qiime2平台,采用dada2算法进行质控,最后基于silva数据库对微生物物种进行注释,获得了10种微生物的分类信息,如表2:
[0029]
表2:微生物物种注释结果
[0030]
微生物编号微生物鉴定结果(属)1fluviicola2sphingomonas3pseudomonas4terrabacter5sphingopyxis6pseudomonas7rhodococcus8brevundimonas
9sphingomonas10pseudomonas
[0031]
(6)结果验证:将由单菌落制备成的10管dna悬液直接送公司逐个进行测序,结果与本方法一致,表明本方法结果准确可靠。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1