一种3-异丁基戊二酸的制备方法
1.本发明涉及普瑞巴林关键中间体的制备领域,具体涉及一种3-异丁基戊二酸的制备方法。具体来说是一种以氰乙酰胺和异戊醛为原料,通过添加硫酸铵改进工艺,适宜工业化生产的3-异丁基戊二酸的制备方法。
背景技术:
2.普瑞巴林,其商品名为乐瑞卡(lyrica),是一种新型γ-氨基丁酸类似物,可用于治疗疱疹后神经痛、外周神经痛、糖尿病性外周神经病引起的疼痛,具有良好的抗癫痫治疗效果。
3.普瑞巴林的制备方法主要有以下四种:
4.一、天然手性库法:采用l-亮氨酸为起始原料,经溴代、酯化、烷基化、水解、环化得(3s)-异丁基内酯,再经碘代、叠氮化、水解、催化氢化得目标产物普瑞巴林。
5.二、不对称合成法:以(e)-n-(5-甲基己-2-烯基)-苯甲酰胺为原料,经选择性加氢,水解脱去苯甲酰胺,最后在二氧化铂的催化下加氢得普瑞巴林。
6.三、酶拆分法:选用异戊醛与丙二酸二乙酯为原料,在有机胺催化下进行脑文格缩合反应、1,4-迈克尔加成反应上一个氰基生成中间体,该中间体经过水解酶或者脂肪酶拆分后,再经过催化氢化等一系列过程得到普瑞巴林。
7.四、化学拆分法:从3-异丁基戊二酸出发得到3-氨甲酰基-5-甲基己酸,该物质可先降解成3-氨甲基-5-甲基己酸,再用(s)-扁桃酸拆分,降解成目标产物普瑞巴林。
8.普瑞巴林在现有工业生产中大多采取化学拆分法,在化学拆分法中,3-异丁基戊二酸是重要的中间体,所以改善3-异丁基戊二酸的制备工艺对普瑞巴林工业生产具有重要意义。
9.现有3-异丁基戊二酸的制备方法,都是以异戊醛和含有活泼氢的羰基化合物为原料,经过脑文格缩合反应、1,4-迈克尔加成反应,然后酸性水解,浓酸下脱羧,萃取得3-异丁基戊二酸。
10.尤海峰等人提出了碱性催化下,以丙二酸二乙酯为含有活泼氢的羰基化合物,与异戊醛经过脑文格缩合反应与脱水反应后得到2-羧乙基-5-甲基-2-己烯酸乙酯。随后加热脱羧得5-甲基-2-己烯酸乙酯,再与丙二酸二乙酯迈克尔加成。最后酸性水解,浓酸下脱羧,萃取得到3-异丁基戊二酸。由于2-羧乙基-5-甲基-2-己烯酸乙酯与丙二酸二乙酯直接加成时加成位阻大,很难反应完全。所以该路线经脑文格缩合、脱水反应得到2-羧乙基-5-甲基-2-己烯酸乙酯后,没有直接与丙二酸二乙酯加成,而是首先脱羧生成空间位阻较小的5-甲基-2-己稀酸乙酯,再与丙二酸二乙酯迈克尔加成。该方法中间体需要提纯分离,反应溶剂、催化剂体系需多次更换,操作步骤繁琐,同时三废量大增,不适合用于工业化生产。
11.张静等人提出了在碱性催化下,以氰乙酸乙酯为含有活泼氢的羰基化合物,与异戊醛经脑文格缩合反应,脱水后再与丙二酸二乙酯迈克尔加成得到2-氰基-4-羧乙基-3-异丁基戊二酸乙酯。然后酸性水解,浓酸下脱羧,萃取得3-异丁基戊二酸。该路线由于使用了
氰乙酸乙酯和丙二酸二乙酯两种活泼氢的羰基化合物,单羧基副产物在产品中含量高,同时该路线反应时间较长,操作较为繁琐。
12.zdenko hamersak和周步高提出碱性催化下,以氰乙酰胺为含有活泼氢的羰基化合物,与异戊醛经脑文格缩合反应。脱水后再与氰乙酰胺迈克尔加成,得2,4-二氰基-3异丁基戊二酰胺。然后酸性水解,再在浓酸下脱羧,萃取得3-异丁基戊二酸。该路线脑文格缩合、脱水、迈克尔加成、酸性水解、浓酸下脱羧一系列反应可以采用“一锅法”完成,而且反应溶剂是水,所以该路线操作方便,节约能耗,且产生三废少,有利于工业化生产。但该路线存在的问题是反应(缩合、脱水和加成在一起)开始不久,反应液中出现大量白色固体,固体将溶剂水和反应液吸湿或包夹,整个反应物料变为膏状物,搅拌无法使反应物料充分混合,反应无法快速充分完成。所以虽然反应本身活化能低,在冰水浴中就能进行,但即使反应进行30多小时,反应还是不充分,产物中仍有单羧基化合物,导致产率和纯度降低无法达到医药中间体含量的要求。
13.针对这一问题,为了改进反应,从而让反应物料充分混合,反应充分完成。zdenko hamersak等人采取加入溶剂的方法,在反应生成大量白色固体导致流动性很差难以搅拌时,加入二氯甲烷以改变其流动性。而周步高等人采取在反应中产生大量膏状物难以搅拌时,将反应升温至70~80℃,直至反应液澄清且搅拌良好。由于膏状物几乎不溶于溶剂,所以加入溶剂并没有真正解决搅拌的问题,而且加入溶剂对后面的水解与脱羧反应均有影响,会产生反应温度难以提升且反应不充分的问题,其最终收率也仅有74%。而升温后副反应增多,且反应副产物大量增加,导致3-异丁基戊二酸的收率与纯度难以提升。
14.cn 106278931 a报道了采用异戊醛与氰乙酰胺为原料,3-异丁基戊二酸单酰胺的制备方法,其中在氰乙酰胺与异戊醛在溶剂中反应制备2,4-二氰基-3异丁基戊二酰胺时,使用过相转移催化剂。实验表明反应生成大量白色固体导致流动性很差难以搅拌,固体粘稠,也无法过滤出来,直接加酸水解后,单羧基化合物含量高。
15.从以上对现有的3-异丁基戊二酸制备路线分析可以看出,现有的3-异丁基戊二酸制备方法存在步骤繁琐,三废多,或反应时间长,单羧基物杂质含量高,反应体系粘稠,难于搅拌的问题。
技术实现要素:
16.本发明的目的在于克服背景技术中指出的已有3-异丁基戊二酸制备方法的不足,提供一种工艺简捷,杂质单羧基化合物含量低,三废少,适用于工业化生产的3-异丁基戊二酸的制备新方法。
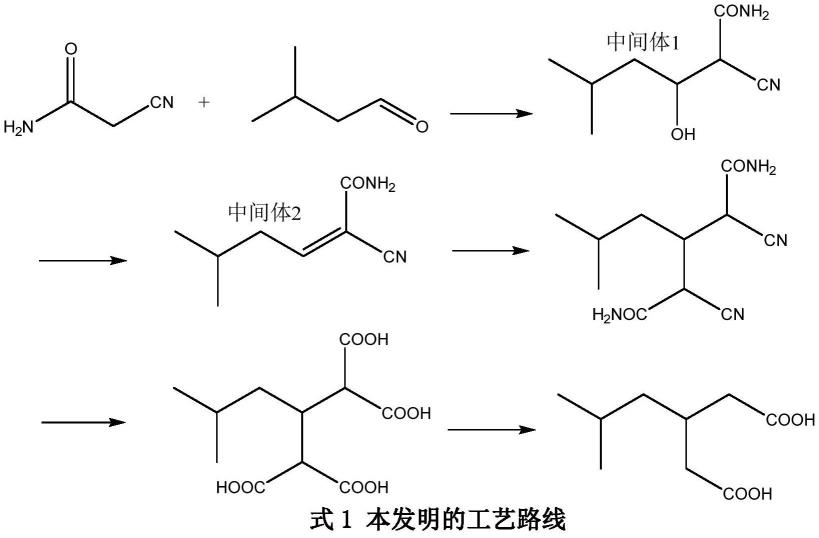
17.本发明目的的实现,主要是以氰乙酰胺与异戊醛为原料,采用如式1路线。
[0018][0019]
具体包括如下步骤:
[0020]
(1)在硫酸铵存在,碱性催化下,氰乙酰胺与异戊醛在溶剂中反应制的2,4-二氰基-3异丁基戊二酰胺;
[0021]
其中,反应温度为0~35℃,应时间为8~25h,氰乙酰胺与异戊醛摩尔比为2.0~3.0。
[0022]
碱性催化剂为吗啡啉等有机碱和无机碱,溶剂为水、二甲苯、甲苯、苯中的一种或几种的混合物。
[0023]
优选地,反应温度为10~20℃,反应时间为10~14h,氰乙酰胺与异戊醛摩尔比为2.0~2.2,溶剂为水。
[0024]
(2)向步骤(1)中加硫酸,水解2,4-二氰基-3异丁基戊二酰胺,生成2,4-二羧基-3-异丁基戊二酸;
[0025]
(3)2,4-二羧基-3-异丁基戊二酸脱羧生成3-异丁基戊二酸;
[0026]
步骤(2)、(3)中,反应温度为70~140℃。
[0027]
步骤(1)、(2)、(3)采用一锅法;
[0028]
步骤(2)、(3)依次,或同时在一个温度段进行;
[0029]
(4)步骤(3)反应液用溶剂萃取,有机溶相脱溶剂结晶得产品3-异丁基戊二酸;
[0030]
溶剂为二甲苯、甲苯、苯中的一种,或几种的混合溶剂。
[0031]
(5)步骤(4)水相通氨气中和,结晶得硫酸铵。
[0032]
步骤(5)所得硫酸铵可以套用在步骤(1),也可以进一步精制为可市售产品。
[0033]
有益效果:本发明方法适用于以氰乙酰胺与异戊醛为原料制备3-异丁基戊二酸。技术方案克服了现有方案难于搅拌的难题,具有工艺简洁,副产物单羧基物含量低,三废少等特点,可以实现工业化生产。
附图说明:
[0034]
图1为实施例7制备的3-异丁基戊二酸的色谱图。
[0035]
图2为实施例8制备的3-异丁基戊二酸的色谱图。
具体实施方式
[0036]
以下结合具体实施例对本技术进行示例性说明,但实施例仅作为例子给出,不视为本发明的全部技术方案,不是对本发明总的技术方案的限定。凡具有相同或相似技术特征,简单改变或替换的,均属本发明保护范围。
[0037]
实施例1
[0038]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入14g氰乙酰胺、0.25g吗啡啉与11.5g硫酸铵水溶液,控温0~10℃。缓慢滴加异戊醛溶液(浓度100%)6.5g,滴完后继续保温反应12h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸0.55mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在100~110℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入二甲苯萃取,萃取相减压蒸除大部分二甲苯,冷却析出产物,产率为85.4%。
[0039]
实施例2
[0040]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入28g氰乙酰胺、0.5g吗啡啉与23g硫酸铵水溶液,控温10~20℃。缓慢滴加异戊醛溶液13g,滴完后继续保温反应12h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸1.1mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在110~120℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入甲苯萃取,萃取相减压蒸除大部分甲苯,冷却析出产物,产率为89.4%。
[0041]
实施例3
[0042]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入28g氰乙酰胺、0.5g吗啡啉与23g硫酸铵水溶液,控温20~35℃。缓慢滴加异戊醛溶液13g,滴完后继续保温反应8h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸1.1mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在120~130℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入甲苯萃取,萃取相减压蒸除大部分甲苯,冷却析出产物,产率为87.3%。
[0043]
实施例4
[0044]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入14g氰乙酰胺、0.25g吗啡啉与11.5g硫酸铵水溶液,控温20~35℃。缓慢滴加异戊醛溶液6.5g,滴完后继续保温反应16h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸0.55mol,滴完后在20~30℃下保温反应5h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在130~140℃下进行脱羧反应10h。反应完毕,冷却至80℃,加入二甲苯萃取,萃取相减压蒸除大部分二甲苯,冷却析出产物,产率为87.7%。
[0045]
实施例5
[0046]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入14g氰乙酰胺、0.25g吗啡啉与11.5g硫酸铵水溶液,控温10~20℃。缓慢滴加异戊醛溶液6.5g,滴完后继续保温反应25h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸0.55mol,滴完后在20~30℃下保温反应5h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在120~130℃下进行脱羧反应10h。反应完毕,冷却至80℃,加入二甲苯萃取,萃取相减压蒸除大部分二甲苯,冷却析出产物,产率为89.5%。
[0047]
实施例6
[0048]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入28g氰乙酰胺、0.5g吗啡啉与23g硫酸铵水溶液,控温0~10℃。缓慢滴加异戊醛溶液13g,滴完后继续保温反应25h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸1.1mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在110~120℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入甲苯萃取,萃取相减压蒸除大部分甲苯,冷却析出产物,产率为90.6%。
[0049]
实施例7
[0050]
将1.5g硫酸铵溶于10ml水中得硫酸铵水溶液;在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入14g氰乙酰胺、0.25g吗啡啉与11.5g硫酸铵水溶液,控温10~20℃。缓慢滴加异戊醛溶液6.5g,滴完后继续保温反应12h。反应结束后,升温至15~30℃。在搅拌下滴加浓硫酸0.55mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在120~130℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入二甲苯萃取,萃取相减压蒸除大部分二甲苯,冷却析出产物,色谱分析如说明书附图1所示,产率为91.8%,重结晶后产品纯度为99.7%。
[0051]
实施例8
[0052]
在配有搅拌、温度计和恒压滴液漏斗的250ml四口烧瓶中依次加入14g氰乙酰胺、0.25g吗啡啉,0.2g十二烷基三甲基氯化铵,控温10~20℃。缓慢滴加异戊醛溶液6.5g,1小时滴完,保温反应10h后出现大量粘稠固体,固体被搅拌叶片拨离开,搅拌叶片无法带动固体,继续反应32小时。升温至15~30℃,在搅拌下滴加浓硫酸0.55mol,滴完后在20~30℃下保温反应3h。然后升温至110~120℃回流6h,随后常压脱除部分水后控制温度在120~130℃下进行脱羧反应7h。反应完毕,冷却至80℃,加入二甲苯萃取,萃取相减压蒸除大部分二甲苯,冷却析出产物,色谱分析如说明书附图2所示,产物中单羧基杂质较多,产物产率为75%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1