一种含双环单萜结构嘧啶胺化合物及其制备方法与应用
1.本发明属于生物医药领域,具体地,涉及一种含双环单萜结构嘧啶胺化合物,该含双环单萜结构嘧啶胺化合物的制备方法与应用。
背景技术:
2.杂环化合物是一类重要的有机化合物,在医药、农药、染料、食品、功能材料等领域有着广泛的应用。在众多的含氮和含硫的多杂环化合物中,六元含氮杂环化合物的数量占了绝大多数,并且具有各种不同的生物活性。嘧啶胺类衍生物芳环中含有2个n原子,环外含有取代氨基,因其具有抑菌、抗病毒、抗过敏和抗肿瘤等功效,在农药、医药领域具有重要应用价值。嘧啶胺类杀菌剂通过抑制真菌体内甲硫氨酸的生物合成和细胞壁降解酶的分泌,从而抑制病菌侵入宿主细胞发挥抑菌作用,对灰葡萄孢引起的各种病害防治效果突出,具有活性高、毒性低、不易耐药等特点。已上市的嘧啶胺抑菌剂,如嘧菌胺、嘧霉胺、嘧螨醚和氟嘧菌胺等,可防治灰霉病、白粉病、黑星病、锈病等多种病害。然而,多数嘧啶胺类化合物因为毒性高、污染大等问题,导致难以进入产业化或市场竞争力不强。
3.诺蒎酮和樟脑因其具有独特的双环结构和环外不饱和羰基,因而化学性质特别活泼,可通过异构化、还原、缩合和酯化等反应合成结构丰富的生物活性物质。例如,蒎烷基噻唑衍生物对由脂多糖所致人脐静脉内皮细胞炎性损伤具有显著的抗炎作用(cn 105646394 a),蒎烷基嘧啶衍生物对人乳腺癌细胞、人肺癌细胞、人肝癌细胞具有一定的抗肿瘤活性(cn103965118 a)。樟脑基缩氨基硫脲类化合物(cn 110551049 a)和樟脑基嘧啶对人多发性骨髓瘤细胞(rpmi-8226)、人乳腺癌细胞(mda-mb-231)和人非小细胞肺癌细胞(a549)具有较好的抑制活性(cn 110551070 a)。因此,利用了蒎烷基和樟脑基结构单元具有良好细胞渗透性、优异的生物相容性和低毒性的特点,设计并制备蒎烷基嘧啶胺类化合物和樟脑基嘧啶胺类衍生物的抑菌活性研究,开发新型结构的绿色、高效、低毒、特异性的抑菌剂对于嘧啶胺类新农药创制具有重要意义。
技术实现要素:
4.针对现有技术中存在的不足,本发明的目的在于探求一种来源于天然产物的诺蒎酮和樟脑为原料,制得具有抑菌活性并且低毒的含双环单萜结构嘧啶胺化合物。
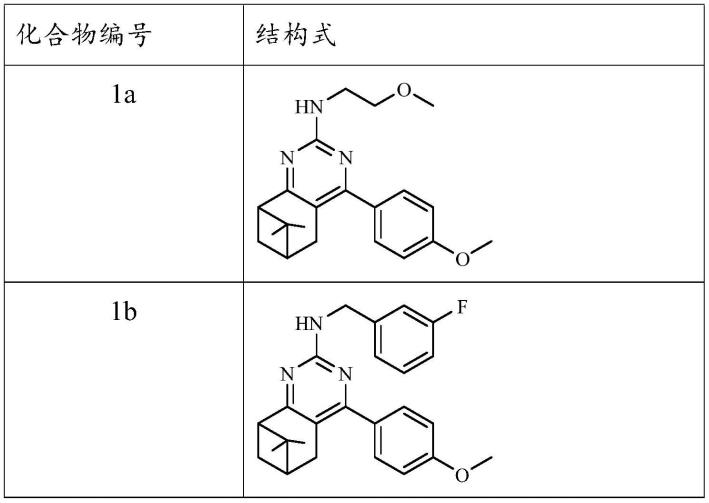
5.为了实现上述目的,本发明提供一种含双环单萜结构嘧啶胺化合物,所述的双环单萜结构嘧啶胺化合物为蒎烷基结构n-取代-4-(4-(二乙氨基)苯基)-7,7-二甲基-5,6,7,8-四氢-6,8-甲基喹唑啉-2-胺和樟脑基结构n-取代-4-(4-甲氧基苯基)-8,9,9-三甲基-5,6,7,8-四氢-5,8-甲基喹唑啉-2-胺,其结构式为式1或式2所示:
[0006][0007]
其中,r1为c
1-c4烷氧基或c
1-c4烷基取代的胺基;r2为碳原子数2-4的烷氧基烷基、苄基或氟取代的苄基。
[0008]
所述c
1-c4烷基包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基。
[0009]
所述c
1-c4烷氧基包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基。
[0010]
所述c
1-c4烷基取代的胺基包括但不限于甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基单取代或多取代的胺基。
[0011]
所述碳原子数2-4的烷氧基烷基包括但不限于甲氧基甲基、甲氧基乙基、甲氧基丙基、乙氧基甲基、乙氧基乙基、丙氧基甲基。
[0012]
所述氟取代的苄基为单氟取代或多氟取代的苄基。
[0013]
根据本发明一种优选实施方式,r1为-och3或-n(c2h5)2;r2为2-甲氧基乙基、苄基、3-氟苄基或2,4-二氟苄基。
[0014]
更优选地,所述含双环单萜结构嘧啶胺化合物选自以下化合物中的一种:
[0015]
[0016][0017]
本发明的另一目的是提供一种含双环单萜结构嘧啶胺化合物的制备方法,反应式如下,烯酮中间体不经分离,具有制备简单、得率较高等优点。
[0018][0019]
具体地,所述的含双环单萜结构嘧啶胺化合物的制备方法,包括如下步骤:
[0020]
(1)以诺蒎酮或樟脑为起始原料,甲醇钠或叔丁醇钾为催化剂,分别与芳香醛衍生物进行羟醛缩合反应,得到3-芳亚甲基蒎烷类化合物或3-芳亚甲基樟脑类化合物,产物不经分离直接与盐酸胍发生环化反应,得到蒎烷基嘧啶类化合物或樟脑基嘧啶类化合物;
[0021]
(2)蒎烷基嘧啶类化合物或樟脑基嘧啶类化合物在氢化钠或叔丁醇钾催化下,于四氢呋喃溶剂中与卤代烃缩合反应,得到式1或式2所示的含双环单萜结构嘧啶胺化合物。
[0022]
根据本发明一种优选实施方式,步骤(1)包括:
[0023]
i)在三口烧瓶中,依次加入诺蒎酮或樟脑、芳香醛衍生物、叔丁醇和叔丁醇钾,开启搅拌器,加热回流反应数小时;
[0024]
ii)反应结束后,反应液加入干燥剂(无水na2so4)搅拌干燥,过滤后,向滤液中加入盐酸胍,加热回流反应数小时;
[0025]
iii)反应结束后,反应液经浓缩除去叔丁醇,再依次经乙酸乙酯萃取、洗涤、干燥、过滤、浓缩后得到蒎烷基嘧啶类化合物或樟脑基嘧啶类化合物的粗产物;
[0026]
iv)粗产物经纯化(重结晶或柱层析),得到蒎烷基嘧啶类化合物或樟脑基嘧啶类化合物。
[0027]
根据本发明一种更具体的实施方式,步骤(1)包括:
[0028]
i)在配有磁力搅拌器、温度计和回流冷凝管的50ml三口烧瓶中,依次加入诺蒎酮或樟脑(14mmol),芳香醛(14mmol)、叔丁醇(30ml)和叔丁醇钾(30~60mmol),开启搅拌器,加热回流反应数小时,至原料转化率达到95%以上(gc跟踪检测);
[0029]
ii)反应液加入na2so4(20mmol)搅拌干燥5min,经过滤后的滤液加入盐酸胍(14mmol),加热回流反应数小时,tlc跟踪检测完成;
[0030]
iii)反应结束后,反应液经浓缩除去叔丁醇,加入乙酸乙酯,分别用蒸馏水和饱和食盐水洗涤至中性,再用无水的na2so4干燥,经过滤、浓缩后得到樟脑基嘧啶类化合物粗产物;
[0031]
iv)粗产物经重结晶纯化,得到蒎烷基嘧啶或樟脑基嘧啶类化合物。
[0032]
根据本发明的方法,所述芳香醛衍生物的结构根据目标化合物的结构确定,例如为对甲氧基苯甲醛、对二乙氨基苯甲醛。
[0033]
根据本发明一种优选实施方式,步骤(2)包括:
[0034]
i)冷阱中控制0℃以内,n2保护下向三口烧瓶中加入thf、蒎烷基嘧啶类化合物或
樟脑基嘧啶类化合物,缓慢分批加入nah,加毕,继续搅拌;
[0035]
ii)反应体系中加入卤代烃,将反应混合物控制在65℃下搅拌数小时,tlc跟踪反应基本完全;
[0036]
iii)反应结束后,冷却至室温,将反应混合物倒入冰水中,用乙酸乙酯萃取,滤液依次经洗涤、干燥、过滤、浓缩、除去乙酸乙酯得到粗品,最后经纯化(柱层析),得到式1或式2所示的含双环单萜结构嘧啶胺化合物。
[0037]
根据本发明一种更具体的实施方式,步骤(2)包括:
[0038]
i)冷阱中控制0℃以内,n2保护下向25ml三口烧瓶中加入thf(10ml)、加入蒎烷基嘧啶或樟脑基嘧啶类化合物(1.0mmol),缓慢分批加入nah(4.0mmol),加毕,继续搅拌30min;
[0039]
ii)反应体系中加入卤代烃(1.1mmol),将反应混合物控制在65℃下搅拌数小时,tlc跟踪反应基本完全;
[0040]
iii)反应结束后,冷却至室温,将反应混合物倒入冰水中,用etoac(2
×
20ml)萃取,滤液分别用蒸馏水和饱和食盐水洗涤至中性,再用无水的na2so4干燥,过滤,经浓缩除去乙酸乙酯得到粗品,最后经柱层析纯化(石油醚/乙酸乙酯=4/1),得到蒎烷基嘧啶胺衍生物或樟脑基嘧啶胺衍生物。
[0041]
根据本发明的方法,本领域技术人员能够根据目标化合物的结构确定所述卤代烃的结构。
[0042]
本发明还有一目的是提供该含双环单萜结构嘧啶胺化合物在抑菌方面的应用。
[0043]
本发明所述含双环单萜结构嘧啶胺化合物可用于制备抗炎化合物,这得益于其具有细菌或真菌抑制活性,所述细菌或真菌包括肺炎克雷伯菌、铜绿假单孢菌、金黄色葡萄球菌、大肠杆菌、耐甲氧西林金黄色葡萄球菌、蜡状芽孢杆菌和白色念珠球菌中的至少一种。
[0044]
本发明所述含双环单萜结构嘧啶胺化合物还具有对单核巨噬细胞白血病细胞的抗炎活性。
[0045]
本发明利用诺蒎酮和樟脑为起始原料,与取代芳醛缩合、盐酸胍环合反应得到蒎烷基嘧啶类化合物和樟脑基嘧啶类化合物,继续与卤代烷取代反应得到蒎烷基嘧啶胺衍生物和樟脑基嘧啶胺衍生物,优点如下:
[0046]
(1)诺蒎酮和樟脑原料廉价易得,来源丰富,有利于工业化生产;
[0047]
(2)合成蒎烷基嘧啶或樟脑基嘧啶类化合物的过程中,中间体无需分离纯化,工艺简单,溶剂消耗少,产率较高,符合清洁工艺与可持续发展的要求;
[0048]
(3)含双环单萜结构嘧啶胺衍生物对细菌和真菌具有显著的抑制活性。生物活性研究结果表明,目标化合物对7种细菌和真菌具有普遍的抑制活性,其中,化合物1a对肺炎链球菌(s.pneumoniae)和大肠杆菌(e.coli)的最低抑制浓度均为1μg/ml,优于阿米卡星;2d对肺炎克雷伯菌(k.pneumoniae)最低抑制浓度为32μg/ml,与阿米卡星相当,对铜绿假单孢菌(p.aeruginosa)最低抑制浓度为16μg/ml,优于阿米卡星。在真菌的抑制活性中,目标化合物1d活性最好,对白色念珠球菌(c.albicans)最低抑制浓度为16μg/ml,与酮康唑相当。此外,抑菌效果较好的1a和2d对小鼠单核巨噬细胞白血病细胞(raw)具有抗炎活性,其中2d(ic
50
=1.87)的活性优于对照品阿司匹林(ic
50
=1.91)。
[0049]
本发明的其它特征和优点将在随后具体实施方式部分予以详细说明。
具体实施方式
[0050]
下面将更详细地描述本发明的优选实施方式。虽然以下描述了本发明的优选实施方式,然而应该理解,可以以各种形式实现本发明而不应被这里阐述的实施方式所限制。
[0051]
实施例1:
[0052]
7,7-二甲基-n-(2-甲氧基乙基)-4-(4-甲氧基苯基)-5,6,7,8-四氢-6,8-桥亚甲基喹唑啉-2-胺(1a)的合成:
[0053][0054]
步骤a:在50ml三口烧瓶中加入30ml叔丁醇,开启磁力搅拌,加入14mmol诺蒎酮,14mmol对甲氧基苯甲醛,58mmol叔丁醇钾,升温至回流,反应6-7h,反应完成,用na2so4干燥,过滤,收集滤液,加入14mmol盐酸胍,升温至回流反应10h。将反应混合物用乙酸乙酯萃取三次(10ml
×
3),合并的有机层用饱和盐水洗涤至中性,用na2so4干燥,浓缩,得到黄色粗产物,柱层析纯化(石油醚/乙酸乙酯=3/1混合溶剂洗脱),得到浅黄色粉末中间体a,收率:86.0%,熔点:173-175℃.1h nmr(400mhz,dmso-d6,ppm)δ:0.65(s,3h),1.22-1.24(m,1h),1.34(s,3h),2.27-2.30(m,1h),2.58-2.60(m,1h),2.61-2.66(m,1h),2.71-2.76(m,1h),2.83-2.87(m,1h),3.80(s,3h),6.21(s,2h),7.00(dd,j=2.8,8.8hz,2h),7.64-7.68(m,2h),
13
c nmr(100mhz,dmso-d6,ppm)δ:175.72,162.40,161.82,160.05,131.50,130.44,113.84,111.99,55.62,50.07,38.61,29.88,29.73,26.08,21.51.hrms-esi:296.1763calcd for c
18h22
n3o[m+h]
+
,found:296.1765.
[0055]
步骤b:冷阱中控制0℃以内,n2保护下向25ml三口烧瓶中加入10ml thf、1.02mmol中间体a、4.06mmol nah,搅拌30min。然后加入1.12mmol 2-溴乙基甲基醚并将反应混合物在65℃下搅拌3h。冷却至室温,将反应混合物倒入冰水中,用etoac(2
×
20ml)萃取。合并的有机层用盐水洗涤,用na2so4干燥,过滤,浓缩得到粗品,经柱层析纯化(pe/ea=4/1)得到类白色固体,收率:79.8%,熔点:125-127℃.1h nmr(400mhz,dmso-d6,ppm)δ:0.69(s,3h),1.23(d,j=9.2,1h),1.36(s,3h),2.29-2.32(m,1h),2.51-2.67(m,2h),2.75-2.91(m,2h),3.26(s,3h),3.46-3.47(m,4h),3.82(s,3h),6.67-6.69(m,1h),7.01-7.04(m,2h),7.01-7.04(d,j=8.4,2h),
13
c nmr(100mhz,dmso-d6,ppm)δ:21.55,26.07,29.78,29.87,38.61,50.25,55.66,58.35,71.20,111.88,113.93,130.48,131.65,160.13,160.60,175.68.hrms-esi:354.2182calcd for c
21h28
n3o[m+h]
+
,found:354.2183.
[0056]
实施例2:
[0057]
n-(3-氟苄基)-4-(4-甲氧基苯基)-7,7-二甲基-5,6,7,8-四氢-6,8-桥亚甲基喹唑啉-2-胺(化合物1b)的制备:
[0058][0059]
参照化合物1a合成方法,得到类白色固体,收率:66.4%.熔点:112-115℃.1h nmr(400mhz,dmso-d6,ppm)δ:7.68(d,j=2.0,6.8hz,2h),7.34-7.39(m,3h),7.09-7.14(m,2h),7.00(dd,j=2.0,7.2hz,2h),4.48-4.50(m,2h),3.81(s,3h),2.79-2.86(m,2h),2.50-2.67(m,2h),2.29-2.30(m,1h),1.35(s,3h),1.23(d,j=9.3,1h),0.68(s,3h).
13
c nmr(100mhz,dmso-d6,ppm)δ:175.79,161.60(d,j=216.3hz),160.27,160.15,137.58(d,j=3.0hz).131.59,130.51,129.67,129.59,115.22(d,j=21hz),113.91,112.14,55.64,50.26,43.93,40.66,29.84,29.81,26.05,21.53.hrms-esi:404.2138calcd for c
25h27
fn3o[m+h]
+
,found:404.2132.
[0060]
实施例3:
[0061]
n-苄基-4-(4-甲氧基苯基)-7,7-二甲基-5,6,7,8-四氢-6,8-桥亚甲基喹唑啉-2-胺(化合物1c)的合成:
[0062][0063]
参照化合物1a合成方法,得到黄色固体,收率:65.1%,熔点:128-130℃.1h nmr(400mhz,dmso-d6,ppm)δ:7.69(d,j=8.8hz,2h),7.34-7.36(m,3h),7.30(t,j=7.5hz,2h),7.00(d,j=8.8hz,2h),4.51-4.53(m,2h),3.80(s,3h),2.79-2.90(m,2h),2.67(t,j=5.5hz,1h),2.57-2.62(m,1h),2.28-2.32(m,1h),1.35(s,3h),1.23(d,j=9.4,1h),0.69(s,3h).
13
c nmr(100mhz,dmso-d6,ppm)δ:175.74,160.61,160.13,141.43,131.61,130.51,128.53,127.73,126.84,113.90,112.03,55.64,50.26,44.61,29.86,29.82,26.06,21.54.hrms-esi:386.2232calcd for c
25h28
n3o[m+h]
+
,found:386.2227.
[0064]
实施例4:
[0065]
n-(2,4-二氟苄基)-4-(4-甲氧基苯基)-7,7-二甲基-5,6,7,8-四氢-6,8-桥亚甲基喹唑啉-2-胺(化合物1d)的合成:
[0066][0067]
参照化合物1a合成方法,得到灰色固体,收率:40.0%,熔点:125-126℃.
[0068]1h nmr(400mhz,dmso-d6,ppm)δ:7.68(dd,j=2.0,6.8hz,2h),7.44(q,j=6.8hz,1h),7.34(t,j=6.6hz,1h),7.15-7.20(m,1h),6.99-7.03(m,3h),4.52-4.53(m,2h),3.81(s,3h),2.80-2.87(m,2h),2.67(t,j=5.6hz,1h),2.51-2.59(m,1h),2.28-2.32(m,1h),1.35(s,3h),1.23(d,j=9.2,1h),0.68(s,3h).
13
c nmr(100mhz,dmso-d6,ppm)δ:175.86,161.59(d,j=256.5hz),162.73,160.54(d,j=258.5hz),161.70,160.39,160.19,131.50,131.16,131.07(d,j=3.0hz),131.00,130.52,124.26(dd,j=15.0,3.0hz),113.92,112.40,111.50(dd,j=20.9,3.6hz),103.86(t,j=25.6hz),55.65,50.25,38.61,37.97(d,j=3.9hz),29.83,26.04,21.52.hrms-esi:422.2044calcd for c
25h26
f2n3o[m+h]
+
,found:422.2046.
[0069]
实施例5:
[0070]
n-苄基-4-(4-(二乙氨基)苯基)-7,7-二甲基-5,6,7,8-四氢-6,8-甲基喹唑啉-2-胺(化合物1e)的合成:
[0071][0072]
参照化合物1a合成方法步骤a,得到黄色白色固体中间体b,收率:78.5%,熔点:146-148℃.1h nmr(400mhz,dmso-d6,ppm)δ:7.63(d,j=8.0hz,2h),6.68(d,j=8.0hz,2h),6.13(s,2h),2.96-2.74(m,2h),2.70-2.54(m,2h),2.40(d,j=81.4hz,2h),1.34(s,4h),1.22(d,j=7.5hz,2h),1.11(t,j=6.0hz,7h),0.67(s,3h).13c nmr(100mhz,dmso-d6,ppm)δ:175.26,162.36,161.64,148.16,130.50,125.35,111.39,110.81,50.07,44.09,38.54,30.29,29.92,26.09,21.49,12.92.hrms-esi:337.2392calcd for c21h29n4[m+h]+,found:337.2390.
[0073]
参照化合物1a合成方法步骤b,中间体b与氯化苄反应,收率:46.5%,熔点:57-60℃.1h nmr(400mhz,dmso-d6,ppm)δ:7.66(d,j=8.6hz,2h),7.28-7.36(m,4h),7.18-7.22(m,2h),6.68(d,j=8.6hz,2h),4.47-4.58(m,2h),3.33-3.40(m,4h),2.88(qd,j=48.9,16.2,3.2hz,2h),2.64(t,j=5.5hz,1h),2.55-2.61(m,1h),2.30-2.33(m,1h),1.35(s,3h),1.22(d,j=9.2hz,1h),1.11(t,j=6.9hz,6h),0.68(s,3h).
13
c nmr(100mhz,dmso-d6,
ppm)δ:174.79,159.97,147.72,141.20,130.05,128.02,127.20,126.31,124.97,110.87,110.34,49.76,44.06,40.08,38.04,29.90,29.41,25.56,21.01,12.44.hrms-esi:427.2862calcd for c
28h35
n4[m+h]
+
,found:427.2877.
[0074]
实施例6:
[0075]
n-(2-甲氧基乙基)-4-(4-甲氧基苯基)-8,9,9-三甲基-5,6,7,8-四氢-5,8-甲氧基喹唑啉-2-胺(化合物2a)的合成:
[0076][0077]
参照1a的合成方法中的步骤a,以樟脑和4-甲氧基苯甲醛为原料,得到浅黄色晶体中间体c,收率:56.8%,熔点:97-98℃.1h nmr(400mhz,dmso-d6,ppm)δ:7.45-7.48(m,2h),7.22(s,1h),6.93-6.96(m,2h),3.86(s,3h),3.11(d,j=4.4hz,1h),2.18-2.22(m,1h),1.76-1.82(m,1h),1.53-1.65(m,2h),1.02-1.05(m,6h),0.83(s,3h).
13
c nmr(100mhz,dmso-d6,ppm)δ:9.31,18.40,20.55,25.92,30.85,46.79,49.23,55.33,57.03,114.18,127.35,128.30,131.39,140.07,160.11,208.27.hrms-esi:271.1698calcd for c
18h23
o2[m+h]
+
,found:271.1700.
[0078]
参照1a的合成方法中的步骤b,中间体c与2-溴乙基甲基醚反应,得到黄色油状物,收率:73.0%.1h nmr(400mhz,dmso-d6,ppm)δ:0.56(s,3h),0.96(s,3h),1.15(s,3h),1.24(m,2h),1.88-2.02(m,1h),2.16-2.18(m,1h),3.08(d,j=3.6hz,1h),3.31(s,3h),3.49-3.50(m,4h),3.82(s,3h),6.70(s,1h),7.05-7.07(m,2h),7.80(d,j=8.8hz,2h).
13
c nmr(100mhz,dmso-d6,ppm)δ:10.04,19.08,20.01,26.02,31.93,41.37,49.97,53.93,55.32,55.60,58.69,71.65,113.76,123.89,129.79,131.15,154.95,160.51,161.08,181.34.hrms-esi:368.2338calcd for c
22h30
n3o2[m+h]
+
,found:368.2397.
[0079]
实施例7:
[0080]
n-(3-氟苄基)-4-(4-甲氧基苯基)-8,9,9-三甲基-5,6,7,8-四氢-5,8-甲氧基喹唑啉-2-胺(化合物2b)的制备:
[0081][0082]
参照化合物1a的合成方法,得到灰色固体,收率:39.1%,熔点:93-96℃.
[0083]1h nmr(400mhz,dmso-d6,ppm)δ:0.54(s,3h),0.96(s,3h),1.16-1.27(m,5h),1.87-1.90(m,1h),2.16-2.17(m,1h),3.07-3.08(m,1h),3.81(s,3h),4.49-4.52(m,2h),7.02-7.05(m,2h),7.09-7.14(m,2h),7.39-7.43(m,3h),7.75-7.78(m,2h),
13
c nmr
(100mhz,dmso-d6,ppm)δ:10.64,19.19,20.19,26.20,32.06,44.24,49.85,53.96,55.45,55.70,114.34,115.19(d,j=21.13hz),123.04,129.78,129.86,130.93,137.72-137.75(d,j=3.0hz),160.25,160.44,161.14-162.65(d,j=151.9hz),181.20.hrms-esi:418.2295calcd for c
26h29
fn3o[m+h]
+
,found:418.2296.
[0084]
实施例8:
[0085]
n-苄基-4-(4-甲氧基苯基)-8,9,9-三甲基-5,6,7,8-四氢-5,8-甲基喹唑啉-2-胺(化合物2c)的合成:
[0086][0087]
参照化合物1a的合成方法,得到黄色固体,收率:44.2%,熔点:114-115℃.1h nmr(400mhz,dmso-d6,ppm)δ:1h nmr(400mhz,dmso-d6):0.55(s,3h),0.96(s,3h),1.24-1.25(m,5h),1.85-1.90(m,1h),2.14-2.18(m,1h),3.07-3.08(m,1h),3.81(s,3h),4.50-4.54(m,2h),7.02-7.05(m,2h),7.18-7.19(m,1h),7.21-7.31(m,2h),7.38-7.39(m,3h),7.75-7.79(m,2h).
13
c nmr(100mhz,dmso-d6,ppm)δ:10.66,19.20,20.20,49.86,53.96,55.70,114.33,122.95,126.84,127.96,128.51,129.86,141.60,160.63(d,j=39.24hz),161.14,162.65,181.20.hrms-esi:400.2389calcd for c
26h30
n3o[m+h]
+
,found:400.2395.
[0088]
实施例9:
[0089]
n-(2,4-二氟苄基)-4-(4-甲氧基苯基)-8,9,9-三甲基-5,6,7,8-四氢-5,8-甲基喹唑啉-2-胺(化合物2d)的合成:
[0090][0091]
参照化合物1a的合成方法,得到灰色固体,收率:43.3%,熔点:59-61℃.1h nmr(400mhz,dmso-d6,ppm)δ:0.54(s,3h),0.96(s,3h),1.16-1.26(m,5h),1.87-1.88(m,1h),2.16-2.17(m,1h),3.07-3.08(m,1h),3.81(s,3h),4.52-4.55(m,2h),7.02-7.05(m,3h),7.15-7.20(m,1h),7.44-7.50(m,2h),7.74-7.77(m,2h).
13
c nmr(100mhz,dmso-d6,ppm)δ:10.02,19.05,19.99,25.91,31.88,39.07(d,j=4.0hz),49.97,54.06,55.35,55.74,103.47(t,j=25.0hz),110.88(dd,j=4.0,17.1hz),113.83,122.85(d,j=4.0hz),124.29,129.83,131.07(dd,j=6.0,7.0hz),159.90,160.55,160.66,161.09(d,j=259.8hz),162.04(d,j=247.5hz),181.73.hrms-esi:436.2200calcd for c
26h28
f2n3o[m+h]
+
,found:436.2203.
[0092]
测试例1:
[0093]
含双环单萜结构嘧啶胺类化合物对肺炎克雷伯菌、铜绿假单孢菌、金黄色葡萄球菌、大肠杆菌、耐甲氧西林金黄色葡萄球菌(mrsa)、蜡状芽孢杆菌和白色念珠球菌抑制活性实验,测试方法如下:
[0094]
以酮康唑和阿米卡星作为抑制真菌和抑制细菌的对照药物,本发明合成的化合物的抑菌活性采用二倍稀释法测试。选用的菌种为肺炎克雷伯菌(k.pneumoniae)、肺炎链球菌(s.pneumoniae)、铜绿假单孢菌(p.aeruginosa)、金黄色葡萄球菌(s.aureus)、大肠杆菌(e.coli)、耐甲氧西林金黄色葡萄球菌(mrsa)、蜡样芽孢杆菌(b.cereus)和白色念珠球菌(c.albicans)。在96孔分析板上,首先将第2孔到第12孔加入100μl纯化水,再将待测化合物1a~1e、2a~2d、阳性对照品酮康唑和阿米卡星用甲醇配成100μg/ml的溶液100μl加入到第1孔,将目标化合物和阳性对照品分别在96孔分析板上进行二倍稀释,从第1到第12孔配成一系列的浓度梯度(1024~1.0μg
·
ml-1
),每孔含100μl溶液,以纯的dmso作为参照,再向每个孔中加入100μl预先配好的菌悬液,充分混匀。最后将96孔分析板置于37℃孵化箱中,细菌培养24h,真菌培养48h,以不产生混浊的最低浓度的孔对应的浓度作为该样品对该测试菌的最低抑菌浓度。每个样品对每种测试菌重复三次,记录实验数据,结果取其平均值,如表1所示。
[0095][0096]
由表1的抑菌结果可知:目标化合物对7种细菌和真菌具有普遍的抑制活性,其中,目标化合物1a对肺炎链球菌(s.pneumoniae)和大肠杆菌(e.coli)的最低抑制浓度均为1μ
g/ml,优于阿米卡星;2d对肺炎克雷伯菌(k.pneumoniae)最低抑制浓度为32μg/ml,与阿米卡星相当,对铜绿假单孢菌(p.aeruginosa)最低抑制浓度为16μg/ml,优于阿米卡星。在真菌的抑制活性中,目标化合物1d活性最好,对白色念珠球菌(c.albicans)最低抑制浓度为16μg/ml,与酮康唑相当。因此本发明所设计的双环单萜嘧啶胺结构衍生物可作为抑菌剂应用。
[0097]
测试例2:
[0098]
采用mtt法检测化合物对raw细胞活力的影响。raw以每孔5
×
104个细胞均匀接种于96孔板中,孵育12小时,使细胞贴壁。10mmol/l的待测化合物母液用无血清培养基稀释为50μm、25μm、12.5μm、6.25μm、3.125μm、1.56μm、0.8μm的含药培养基。弃细胞上清,将含药培养基加入到96孔板中,阳性对照组和阴性对照组均加入相同体积的培养基,继续孵育12小时。弃细胞上清液,加入5μg/ml的lps无血清培养基,继续培养24小时。随后,于酶标仪上检测每孔在492nm下的吸光度,所得数据均用spss软件计算细胞存活率,结果如表2所示。
[0099]
表2化合物对raw的抗炎活性
[0100][0101]
由表2结果可知,化合物1a和2d对raw细胞均有明显的抗炎活性,其中化合物2d(ic
50
=1.87)的活性优于对照品阿司匹林(ic
50
=1.91)。
[0102]
以上已经描述了本发明的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1