基于宏基因组和宏转录组识别油藏驱油功能微生物的方法
1.本发明涉及微生物检测技术领域,尤其是涉及一种基于宏基因组和宏转录组识别油藏驱油功能微生物的方法。
背景技术:
2.我国稠油资源量约为198.7亿吨,年产量高达3087万吨(2017年),已占原油总产量的16.2%。由于稠油粘度高(50-10000mpa.s),在油藏中流动性差,一般以蒸汽热采方法为主,而热采法耗能大、成本高、开采效果差。此外,油藏是一个天然生物反应发生器,同时蕴含了具有各种功能的好氧及厌氧微生物。通过利用微生物来采油的技术绿色环保、成本低,可用于稠油开采,其主要机理是通过微生物在油藏中乳化原油、产气、产表面活性剂、产多糖和降解烃等五方面功能降低稠油粘度、提高水驱效率。
3.近20年来,对油藏环境中微生物的研究已经从最初的纯菌分离培养模式过渡到了依赖测序的分子生物学研究方式。其中,测序手段可以大致分为依赖pcr扩增的测序技术和不依赖pcr扩增的宏基因组与宏转录组测序技术。前者以16s rrna基因克隆文库方法为代表,在油藏环境样品中已经受到广泛应用,通过设计特异性的引物可以扩增出样品中的相关基因序列,从而在基因的水平上阐述微生物的潜在代谢功能。而后者不需要pcr扩增,可以同时测定样品中所有基因的序列信息。因此,采用宏基因组测序分析油藏环境样品可以深入解析样品中潜在的代谢网络,进一步将宏基因组技术和宏转录组技术结合能够得到代谢途径上各个基因的转录水平,从而推断油藏环境下的各种微生物代谢过程。
4.现有的宏基因组学分析手段(例如一种基于宏转录组学和宏基因组学的环境中抗生素抗性基因的活性定量及宿主鉴定方法,申请号202110740585.0)已经可以根据需求对一些常规环境样品的目标功能基因和重要微生物进行分析。但是地下油藏作为一个以厌氧条件为主的特殊环境,如果不对样品的采集和提取过程进行针对性的处理,样品中的微生物组成极易受到干扰而发生变化,rna也会发生降解,从而导致后续的分析无法获得真正的油藏原位微生物数据。并且油藏中微生物的功能多种多样,其中值得关注研究的种类繁多,单一数据库无法对这些功能进行有效的分析,因此必须结合多个公开数据库以及本地自建数据库才能更全面地注释和分析样品中的这些关键功能。
5.可见,基于目前的技术空白,亟需开发一种特别针对油藏环境而开发的基于宏基因组学和宏转录组学的微生物识别、分析方法。
技术实现要素:
6.本发明的目的就是为了解决对现有技术中油藏微生物检测手段可以获得的信息有限的问题,提供了一种联合应用宏基因组和宏转录组准确地识别油藏中微生物和代谢功能的方法。
7.本发明的目的通过以下技术方案实现:
8.本发明的目的是提供一种基于宏基因组和宏转录组识别油藏驱油功能微生物的
方法,包括以下步骤:
9.s1:自油藏产出水样提取总dna和总rna;
10.s2:对获得的总dna和总rna进行测序,获取油藏样品的宏基因组和宏转录组原始数据;
11.s3:通过对宏基因组和宏转录组结果进行分析,识别具有驱油功能的微生物。
12.进一步地,s1中,在提取总dna和总rna之前,在待提取rna的样品中加入抑制剂以抑制厌氧微生物的rna降解。
13.进一步地,s2中,还包括:对油藏样品的宏基因组和宏转录组原始数据进行预处理,得到去除接头和低质量片段的目标数据;
14.所述预处理的过程包括:
15.利用fastp软件分别对对油藏样品的宏基因组和宏转录组中各dna或rna链序列双端的原始数据进行滑窗质量剪裁,同时,根据序列首尾两端的引物信息,利用cutadapt软件去除引物,得到质控后的双端序列数据。
16.进一步地,滑窗质量剪裁的参数为-w 4,-m 20,即滑动窗大小为4,平均质量值为20。
17.进一步地,s3中,对宏基因组和宏转录组结果进行分析的过程包括:
18.对质控后的双端序列数据进行组装、分箱并评估质量,去除冗余后提取其中高质量的mags(宏基因组组装基因组)数据集;
19.根据构建的参考数据库对高质量的mags组装数据进行注释,识别出具有不同驱油功能的功能基因和mags,并计算对应的测序深度和相对丰度;
20.对高质量的mags数据集做进化关系分析,进而做出驱油微生物的群落结构分析;
21.将宏转录组测序得到的序列短片段质控过滤后与mags数据对比,计算出各个基因的转录水平。
22.进一步地,s3中,组装、分箱并评估质量的过程包括:
23.组装:使用拼接程序spades在meta模式下,将质控后的双端序列数据样品短序列拼接成长度不一的contigs(交替片段产物),然后根据双端测序的信息将不同contigs连接成有测序缺口的scaffolds(骨架序列);
24.分箱:采用bowtie2软件将质控后的mags短序列数据信息比对到长序列信息上,获得不同长序列的测序覆盖度信息,进一步同时采用maxbin2、metabat2、concoct三种binning手段从mags中分离出优势菌的基因组,并进一步导入das_tool程序中进行评估,最终整合并提高不同方法生成基因组的质量;
25.评估质量:利用drep软件将质控后的mags中相似度较高的基因组去除冗余,通过checkm工具根据基因组中单拷贝标记基因的有无和数量来估算基因组的完整度和污染度。
26.进一步地,s3中,对高质量的mags组装数据进行注释的过程包括:
27.使用prodigal程序(预测开放阅读框)将拼接后的长序列翻译成编码蛋白序列(cds),并提交到kegg数据库,采用ghostkoala工具进行功能性注释并获得代表不同旁系同源亚基的ko号;
28.同时采用本地软件kofamkoala根据各个ko旁系同源家族蛋白的隐马可夫模型(hmm)和推荐的置信标准给各个蛋白序列注释ko号;
29.最后采用eggnog emapper 2工具给蛋白序列注释cog号,再转换成ko号,使得最终每个蛋白质的ko号注释采用以下顺序:1)ghostkoala ko,2)kofamkoala ko,3)eggnog emapper ko。
30.进一步地,s3中,所述驱油功能包括烷烃降解、产气、产乳化剂、产表面活性剂、产多糖中的一种或多种。
31.进一步地,s3中,识别出具有不同驱油功能的功能基因和mags,并结合bowtie2软件对比得到的各序列的测序覆盖度信息计算对应的测序深度和相对丰度的过程包括:
32.针对氢气还原酶,首先通过本地的氢气还原酶亚组的hmm模型比对找出潜在的功能基因蛋白;
33.之后将潜在的功能基因蛋白序列提交到hyddb数据库的在线分析软件进一步划分氢气还原酶的亚型;
34.针对基因组中潜在的编码次级代谢产物的功能,通过将基因组序列提交至antismash网站,结合不同的工具找到基因组中潜在编码次级代谢产物的基因组,并预测代谢产物的类型;
35.针对数据库中信息缺少的功能基因,单独构建本地的蛋白序列数据库,通过blasp(blast protein)比对方法找到最相似的蛋白序列并进一步分析,所述数据库中信息缺少的功能基因包括厌氧烃降解初始活化基因assa、ebda、ahya、anca,和细菌微室蛋白簇基因中的一种或多种。
36.进一步地,s3中,对高质量的mags数据集做进化关系分析的过程包括:
37.首先将目标序列以及数据库中的相似参比序列下载并合并文件,合并后的序列首先在mafft上排列整齐,并以80%的阈值来选择保守位点,进而使用iq-tree(该算法采用最大似然法构建系统发育树)来两两比对序列并生成最大似然进化树。
38.进一步地,s3中,将宏转录组测序得到的序列短片段质控过滤后与mags数据对比,计算出各个基因的转录水平的过程包括:
39.采用bowtie2软件将高质量的mags数据集中的cdna短片段比对到通过s3中组装分箱后的宏基因组拼接得到的dna长片段上,计算出各个基因的转录水平,以tpm值(transcripts per million)来表示。
40.与现有技术相比,本技术方案的优势在于:
41.本技术方案是一种特别针对油藏环境而开发的基于宏基因组学和宏转录组学的微生物分析方法,整体过程中,自油藏产出水样提取总dna和总rna,测序获取油藏样品的宏基因组和宏转录组原始数据,通过对宏基因组和宏转录组结果进行分析,识别具有驱油功能的微生物,整体过程不依赖传统的速度较慢的微生物单菌分离鉴定手段,适合处理未知物种较多的样本,且可检测极低丰度物种,检测全面、快速。
具体实施方式
42.下面结合具体实施例对本发明进行详细说明,但绝不是对本发明的限制。本技术方案中如未明确说明的软件/程序名称、控制方法、算法等特征,均视为现有技术中公开的常见技术特征。
43.本发明的油藏驱油功能微生物识别方法基于宏基因组和宏转录组测序经行检测,
technologies,美国)和agilent 4200(agilent,加拿大)共同评估了文库的质量。最后在illumina hiseq x-ten平台上,对文库进行了测序,得到了150bp的双端碱基序列。
60.(3)数据分析
61.原始数据通过使用fastp进行质控和过滤。宏基因组数据通过spades经行组装,maxbin2、metabat2和concoct进行分箱。获得的基因组导入das_tool后获得高质量的基因组。使用prodigal将长片段翻译成编码蛋白序列cds,采用ghostkoala,kofamkoala和eggnog emapper 2三种工具给蛋白序列注释,通过hyddb数据库antismash分析基因组的产氢气能力和次级代谢产物合成能力,通过自建本地数据库分析厌氧烃降解能力。
62.宏转录组数据通过bowtie2比对到宏基因组拼接的dna长片段上,计算出各个基因的转录水平tpm。
63.结合样品中基因组的相对丰度和对应的代谢通路可以准确地识别油藏中的主要微生物和代谢功能,再结合转录组数据可以得到这些微生物的表达活性。
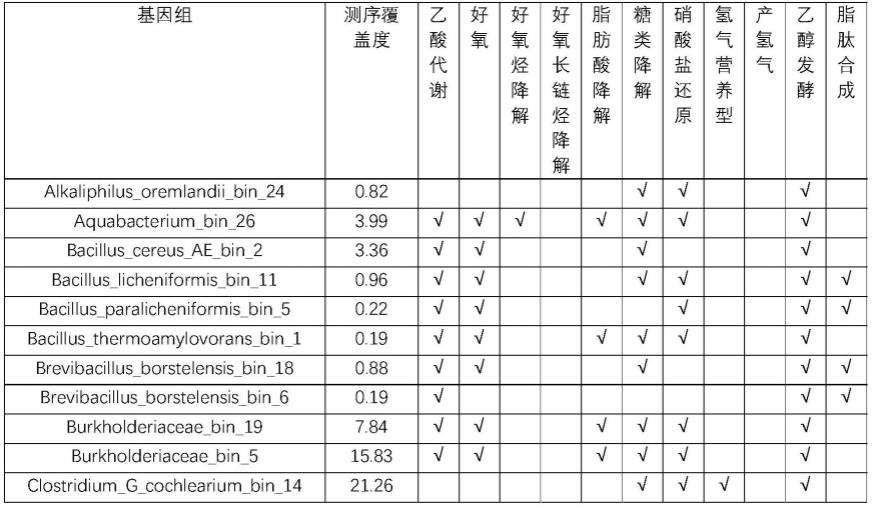
64.以大庆油田的一份产出液样品为例,表1中的数据为各基因组测序覆盖度和代谢通路。从表中可以得知该样品中最主要的微生物为变形菌门的kerstersia_gyiorum和serratia_nematodiphila以及厚壁菌门的enterococcus。样品中的微生物大都为具有硝酸盐还原能力的兼性厌氧微生物,且样品中存在针对不同链长的好氧烃降解菌。近一半的微生物都具有完整的合成脂肽的代谢通路。此外,大多数氢营养型微生物同时具有产氢能力。以上这些信息可以为油藏提高采收率和防腐蚀方面的决策提供理论依据。
65.基因组信息表1
66.[0067][0068]
上述的对实施例的描述是为便于该技术领域的普通技术人员能理解和使用发明。熟悉本领域技术的人员显然可以容易地对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中而不必经过创造性的劳动。因此,本发明不限于上述实施例,本领域技术人员根据本发明的揭示,不脱离本发明范畴所做出的改进和修改都应该在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1