一种多孔配位聚合物及制备和对同位素体的分离方法
1.本发明涉及同位素体分离技术领域,具体涉及了一种多孔配位聚合物及制备和对同位素体的分离方法。
背景技术:
2.同位素体是化学组成完全相同,仅在同位素组成上不同的物质,例如氢同位素d2/h2,水的同位素体d2o/hdo/h2o/h
218
o等。发展有效的方法将同位素体进行识别和分离,无论在基础研究还是工业应用方面都是极其重要的,然而至今仍是化学领域的一大挑战。为了将同位素广泛的应用于工业、生物医疗和科学研究,化学家们试图用分子化学的方法区分同位素,例如使用包括笼状化合物或刚性多孔材料在内的主体材料捕获其中一种同位素。然而,由于同位素体具有相同的化学结构和尺寸,热力学性质又极其相似,因此将同位素体相互分离十分困难,其中水的同位素体又是最难区分的。水(h2o)和重水(d2o)作为水的两种典型的同位素体,它们的物理化学性质,如熔点(273.15k vs.276.94k)、沸点(373.15k vs.374.56k)、键能(458.9kj mol-1 bond-1 vs 466.4kj mol-1 bond-1)等都非常相似,导致工业上采用的高温多级精馏技术和电解水技术分离水同位素体的效率极低(分离因子1.02~2.0)。此外,当h2o和d2o混合时,通过质子交换化学平衡,将快速生成半重水(hdo)。在298k时,平衡常数k=[hdo(liquid)]2/{[h2o(liquid)]
·
[d2o(liquid)]}=3.85,三者始终共存。因此,工业上采用的geib-spevack法等质子交换平衡的热力学方法分离水的同位素体的效率也极低(分离因子1.2~2.0)。此外,h2o和d2o的分子动力学直径非常小且完全相同这使得利用主体材料进行吸附分离也相当困难。在过去的几十年里,化学家们一直尝试利用多孔材料中吸附性能的差异实现水的同位素体分离,然而至今未见一例成功的报道。
[0003]
另一方面,氢同位素可以在小于40k的低温下通过动力学量子筛分效应实现有效分离,这是因为氢同位素在低温下扩散速率略有差异,通过严格控制多孔材料的微孔孔径等于d2的德布罗意波长时,可产生量子隧穿效应,从而可以放大这种差异,而水的同位素体即使在低温下扩散速率也没有表现出明显的差异,从而实现有效分离。迄今为止,控制水的同位素体扩散的材料还未曾报道。
技术实现要素:
[0004]
本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明提出一种多孔配位聚合物及其制备方法和应用,本发明中的配位聚合物具有可以翻折运动的翻折单元,配位聚合物内部独特的孔径结构及通道允许水的同位素体通过并控制其扩散,且水的同位素体在本发明中的配位聚合物中的扩散速率不同,从而实现了对水的同位素体的分离,且298k下,本发明中的配位聚合物从h2o/hdo/d2o三元混合体系中动态分离h2o的分离系数高达约210;此外,本发明中的配位聚合物在水和有机溶剂中都具有较高的稳定性,从而保证了水同位素体分离的可实施性。
[0005]
本发明的第一个方面,提供了一种配位聚合物,所述配位聚合物是由金属离子和有机配体通过配位键自组装而成。
[0006]
根据本发明第一方面的内容,在本发明的一些实施方式中,所述有机配体具有式(ⅰ)或式(ⅱ)中的通式:
[0007][0008]
在本发明的一些优选实施方式中,所述r1~r6分别独立地选自-h、-o、-ch3、-c2h5、-och3中的一种。
[0009]
在本发明的一些优选实施方式中,所述r7~r
14
分别独立地选自-h、-o、-ch3、-c2h5、-och3中的一种。
[0010]
在本发明的一些优选实施方式中,所述有机配体中间苯二甲酸5-位上是由亚氨基芪、亚氨基二苄、5h-二苯并[b,f]氮杂环庚烯-10(11h)-酮、10-甲氧基亚氨基芪中的至少一种取代。
[0011]
在本发明的一些优选实施方式中,所述有机配体中包含可以翻折运动的单元。
[0012]
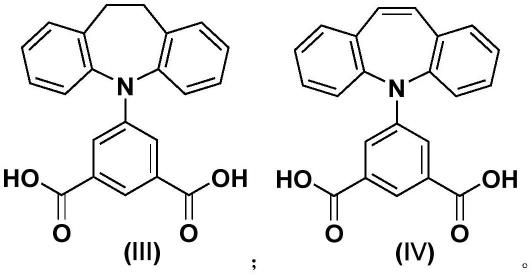
在本发明的一些更优选实施方式中,所述有机配体具有式(ⅲ)或式(ⅳ)中的结构:
[0013][0014]
在本发明的一些优选实施方式中,所述金属离子选自钴、锌、镍、铜、钠、钾、钙中的一种。
[0015]
在本发明的一些更优选实施方式中,所述金属离子是来自可溶性的金属盐。
[0016]
在本发明的一些更优选实施方式中,所述金属盐选自硝酸钴、硝酸锌、硝酸镍、硝酸铜、硝酸钠、硝酸钾、硝酸钙、醋酸铜、醋酸钾、醋酸钴、醋酸锌、硫酸铜、硫酸锌、硫酸钠、硫酸钴、硫酸锌、氯化铜、氯化锌、氯化钴、氯化镍、氯化钙中的一种。
[0017]
在本发明的一些优选实施方式中,所述配位聚合物的配位数为2。
[0018]
在本发明的一些优选实施方式中,所述配位聚合物为多孔配位聚合物。
[0019]
在本发明的一些更优选实施方式中,所述配位聚合物的孔径为且所述孔径具有动态调节能力。
[0020]
本发明的第二方面,提供了一种本发明第一方面所述的配位聚合物的制备方法,所述配位聚合物通过反应得到的有机配体与含有金属离子的盐通过配位键自组装而成,其中x为卤素原子。
[0021]
根据本发明第二方面的内容,在本发明的一些实施方式中,所述金属离子是来自可溶性的金属盐。
[0022]
在本发明的一些优选实施方式中,所述金属盐选自硝酸钴、硝酸锌、硝酸镍、硝酸铜、硝酸钠、硝酸钾、硝酸钙、醋酸铜、醋酸钾、醋酸钴、醋酸锌、硫酸铜、硫酸锌、硫酸钠、硫酸钴、硫酸锌、氯化铜、氯化锌、氯化钴、氯化镍、氯化钙中的一种。
[0023]
在本发明的一些更优选的实施方式中,所述有机配体由以下步骤制得:
[0024]
s1.将2-二环己基磷-2
′
,4
′
,6
′‑
三异丙基联苯、碳酸铯和三(二亚苄基丙酮)二钯(0)置于容器中,在惰性气体的保护下加入溶剂进行反应;
[0025]
s2.当完全消失时停止反应,加入乙酸乙酯和硅藻土,滤掉固体后得到的有机相进行萃取;
[0026]
s3.收集有机相后进行柱层析分离、洗脱后得到化合物1s3.收集有机相后进行柱层析分离、洗脱后得到化合物1
[0027]
s4.将化合物1溶于有机溶剂后加入naoh水溶液进行回流反应;
[0028]
s5.冷却后除掉有机溶剂,调节ph值,滤出固体后洗涤干燥后得到有机配体。
[0029]
在本发明的一些优选实施方式中,步骤s1中所述的溶剂为超干甲苯。
[0030]
在本发明的一些优选实施方式中,步骤s1中所述反应的温度为110~120℃。
[0031]
在本发明的一些优选实施方式中,步骤s1中所述反应的时间为14~18h。
[0032]
在本发明的一些优选实施方式中,步骤s2中采用薄层色谱法对反应进行监测。
[0033]
在本发明的一些优选实施方式中,步骤s2中萃取的顺序为使用乙酸乙酯萃取3次,使用饱和食盐水萃取3次。
[0034]
在本发明的一些优选实施方式中,步骤s3中在柱层析分离前所述有机相进行干燥处理。
[0035]
在本发明的一些优选实施方式中,所述干燥处理的方法可以是使用无水硫酸镁进行干燥。
[0036]
在本发明的一些优选实施方式中,步骤s3中洗脱的洗脱剂为乙酸乙酯/正己烷。
[0037]
在本发明的一些更优选实施方式中,所述乙酸乙酯和正己烷的体积比为(3~6):1。
[0038]
在本发明的一些优选实施方式中,步骤s3中化合物1的产率为55~60%。
[0039]
在本发明的一些优选实施方式中,步骤s4中的有机溶剂为四氢呋喃(thf)和甲醇(meoh)的混合物。
[0040]
在本发明的一些更优选实施方式中,thf和meoh的体积比为1:1。
[0041]
在本发明的一些优选实施方式中,步骤s4中的naoh水溶液的浓度为1~3m。
[0042]
在本发明的一些优选实施方式中,步骤s4中的回流反应的时间为14~18h。
[0043]
在本发明的一些优选实施方式中,步骤s5中除掉有机溶剂的方法包括旋转干燥法。
[0044]
在本发明的一些优选实施方式中,步骤s5中的ph值为1~2。
[0045]
在本发明的一些更优选实施方式中,使用冰盐酸进行ph值调节。
[0046]
在本发明的一些优选实施方式中,步骤s5中使用水洗涤固体2~4次。
[0047]
在本发明的一些优选实施方式中,步骤s5中干燥的温度为50~70℃。
[0048]
在本发明的一些优选实施方式中,步骤s5中干燥的时间为10~14h。
[0049]
本发明的第三方面的内容,提供了本发明第一方面所述的配位聚合物在水的同位素体分离中的应用。
[0050]
根据本发明第三方面的内容,在本发明的一些实施方式中,所述配位聚合物使用前需进行活化。
[0051]
在本发明的一些优选实施方式中,所述活化的方法为在353k~393k的温度下真空静置8~15h。
[0052]
其中,适量高的温度是为了使得本发明的配位聚合物孔道中的溶剂充分挥发,从而可以提高配位聚合物对水的吸附量,对水的同位素体实现更有效的分离。
[0053]
在本发明的一些优选实施方式中,所述配位聚合物在278k~473k的温度内进行分离。
[0054]
在本发明的一些优选实施方式中,所述配位聚合物在水中的吸附量为50~70ml/g。
[0055]
在本发明的一些优选实施方式中,所述水的同位素体包括d2o、hdo、h2o、h
218
o。
[0056]
在本发明的一些优选实施方式中,所述配位聚合物用来分离两种或两种以上的水的同位素体。
[0057]
发明人提出一种低耗能的“扩散控制”策略,实现有效的分离水的同位素体。该策
略的本质是利用动态“局域柔性”框架材料控制水的同位素体的扩散,并放大水的同位素体的扩散速率差异。动态“局域柔性”框架材料会根据动力学差异从水的同位素体混合物中优先吸附一种同位素体分子,从而实现在动力学上对水的同位素体的有效识别。具体的方法是,建立了一个通过配体基团翻折运动来控制水的同位素体分子扩散过程的“局域柔性”多孔配位聚合物体系。在刚性骨架的多孔配位聚合物(pcps)的笼状孔壁上编入温度响应的动态“开关”表现出温度响应的吸附行为,能够放大水的同位素体之间扩散速率的细微差异,实现对h2o的高选择性吸附。在298k下本发明中的配位聚合物从h2o/hdo/d2o三元混合体系中动态分离h2o的分离系数高达约210。
[0058]
本发明的有益效果是:
[0059]
(1)本发明中的配位聚合物具有可以翻折运动的翻折单元,配位聚合物允许水的同位素体通过并控制其扩散,且水的同位素体在本发明中的配位聚合物中的扩散速率不同,从而实现了对水的同位素体的分离,298k下本发明中的配位聚合物从h2o/hdo/d2o三元混合体系中动态分离h2o的分离系数高达约210。
[0060]
(2)本发明中的配位聚合物在水和各种有机溶剂中都具有较高的稳定性,无论是在常温还是高温浸泡后,本发明中的配位聚合物的粉末x射线衍射分析(pxrd)谱图都未发生明显的变化,从而保证了水同位素体分离的可实施性。
附图说明
[0061]
图1为本发明实施例2中的有机配体idb-ipa的氢谱图;
[0062]
图2为本发明实施例2中的有机配体idb-ipa的碳谱图;
[0063]
图3为本发明实施例2中的有机配体idb-ipa的质谱图;
[0064]
图4为本发明实施例1的有机配体中dbap环和本发明实施例2的有机配体中idb环的翻折运动势能曲线;
[0065]
图5为本发明实施例中所制备的配位聚合物的光学显微镜照片;
[0066]
图6为fdc-1a和fdc-2a的活化晶体结构;
[0067]
图7为本发明实施例中所制备的配位聚合物在不同溶剂中浸泡7天后的pxrd谱图;
[0068]
图8为298k下本发明实施例中的配位聚合物对h2o和d2o的吸-脱附曲线以及时间依赖吸附曲线;
[0069]
图9为不同温度下本发明实施例中的配位聚合物吸附h2o和d2o的压力-扩散速率-吸附量全景图;
[0070]
图10为298k温度下本发明实施例中的配位聚合物对水同位素体分离的麦凯布-蒂勒图;
[0071]
图11为298k温度下本发明实施例中fdc-1a和fdc-2a的吸附相中h2o分离因子随原料蒸汽中h2o含量的变化情况。
具体实施方式
[0072]
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述而更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进
行修改或替换,但这些修改和替换均落入本发明的保护范围内。
[0073]
实施例1有机配体的合成
[0074]
实施例1中的有机配体的合成路线为:
[0075][0076]
具体的制备步骤为:
[0077]
将5-碘间苯二甲酸二甲酯(19.20g,60.0mmol,1.2当量)、亚氨基芪(9.66g,50.0mmol,1.0当量)、2-二环己基磷-2
′
,4
′
,6
′‑
三异丙基联苯(xphos,1.19g,2.5mmol,0.05当量)、碳酸铯(cs2co3,32.58g,100.0mmol,2.5当量)和三(二亚苄基丙酮)二钯(0)(pb2(dba)3,1.37g,1.5mmol,0.03当量)置于500ml圆底烧瓶中,排空烧瓶中的空气并以氩气填充,在氩气保护下,加入200ml超干甲苯(toluene),115℃搅拌16h。采用薄层色谱法(tlc)监测反应,当原料亚氨基芪完全消失时停止反应并冷却至室温,在烧瓶里加入50ml乙酸乙酯和硅藻土,搅拌后滤掉固体,有机相用乙酸乙酯(200ml
×
3次)和饱和食盐水(100ml
×
3次)进行萃取。收集有机相,使用无水硫酸镁干燥后旋干上样进行柱层析分离,乙酸乙酯/正己烷(体积比为(3~6):1)洗脱,80℃真空干燥12h,得到粉末其质量为11.18g,产率为58%。将含有化合物(5.0g,12.95mmol)的thf/meoh(100ml,1/1v/v)溶液加入500ml圆底烧瓶中,然后加入2m的naoh水溶液(100ml,200mmol),回流反应16h。冷却至室温后,旋转蒸发除掉有机溶剂,再用3%的冰盐酸调节溶液ph至1-2,滤出固体,用去离子水洗涤三次。60℃真空干燥12h,得到白色粉末记为dbap-ipa,其质量为9.1g,产率为98%。
[0078]
实施例2有机配体的合成
[0079]
实施例2中的有机配体的合成路线与实施例1相似,区别在于将化合物替换为
[0080]
具体的合成路线为:
[0081][0082]
所得到的有机配体记为idb-ipa,其质量为9.1g,产率为98%。
[0083]
图1~图3分别是翻折运动有机配体idb-ipa的氢谱、碳谱及质谱图,从图中可以看出,本发明实施例中制备了可以翻折运动的有机配体idb-ipa,同时也计算了本发明实施例1和2中的有机配体dbap-ipa和idb-ipa中翻折能垒与二面角(c1n1c2c3)的关系,其中二面角(c1n1c2c3)的范围是从100
°‑
160
°
(10
°
间隔),如图4所示,其中,图4a为有机配体dbap-ipa中dbap环的翻折能垒和二面角c1n1c2c3的关系,图4b为有机配体idb-ipa中idb环的翻折能垒和二面角c1n1c2c3的关系。从图4中可以看出,无论是dbap环还是idb环,其翻折运动的能量变化(δe)都很小,相对于二面角20
°
的变化δe小于20kj mol-1
。上述结果表明,本发明实施例中的有机配体中的dbap环或idb环翻折时的能量变化较小,在很低的温度下都可以有效的发生,并且随温度的升高翻折运动的振幅提高。
[0084]
实施例3配位聚合物的合成
[0085]
在30℃下将500mg(1.40mmol)实施例1中的dbap-ipa溶于50ml的二甲基乙酰胺(dma)中,然后加入676mg(2.80mmol)的cu(no3)2·
3h2o水溶液(50ml)。将混合液在80℃烘箱加热24h,得到尺寸接近1mm的墨绿色块状单晶,如图5a所示,过滤晶体,用dma和水分别洗涤三次,30℃下晾干,得到cu(dbap)(记为fdc-1,538mg,产率78%)。
[0086]
实施例4配位聚合物的合成
[0087]
在30℃下将500mg(1.40mmol)实施例2中的idb-ipa溶于50ml的dma中,然后加入676mg(2.80mmol)的cu(no3)2·
3h2o水溶液(50ml)。将混合液在80℃烘箱加热24h,得到尺寸接近1mm的墨绿色块状单晶,如图5b所示,过滤晶体,用dma和水分别洗涤三次,30℃下晾干,得到cu(idb)(记为fdc-2,538mg,产率80%)。
[0088]
当然,本领域的技术人员可以按照上述步骤合成其他的有机配体,并与不同的金属离子通过配位键进行自组装,制备得到其他的配位聚合物。
[0089]
fdc-1和fdc-2在水同位素体分离中的应用
[0090]
一、实验流程
[0091]
1.装样及活化:将实施例3或4中的配位聚合物(0.40g)装入圆柱形样品池(直径为8mm)中,在393k温度下真空干燥活化10h,然后将样品池安装在bel-cat ii仪器上,并在仪器上原位活化1h,活化过程中以10sccm的恒定速率吹扫氦气1h;活化过程需确保样品吸附的溶剂完全去除,活化后的样品分别记为fdc-1a和fdc-2a。
[0092]
2.吸附过程:以10k min-1
的速率从393k降温到298k,当温度达到指定温度时,混合蒸汽以10sccm的速率吹过样品(混合蒸汽流速由质量流量计控制),吸附过程中,温度始终保持在298k。
[0093]
3.排出样品池内未吸附的蒸汽:在298k的温度下,以10sccm的流速连续吹扫氦气1h,将样品池和管路中的多余蒸汽排出。
[0094]
4.吸附气的脱附与分离检测:以10k min-1
的速度将样品池温度升高至393k,这时样品内部吸附的蒸汽进行脱附,同时以10sccm的速度连续吹扫氦气将脱附的蒸汽输送到检
测器,用质谱检测脱附的水蒸汽。通过对扣除基线后的电流信号面积进行积分,计算释放出的h2o、hdo和d2o的比值。
[0095]
本领域的技术人员也可以根据实际的需要选择不同的活化温度,并不局限于393k。
[0096]
本领域的技术人员也可以根据实际需要选择吸附的温度,例如,吸附的温度也可以为278k、288k、308k、323k或393k。
[0097]
其中,h2o分离因子定义为:
[0098][0099]
式中,x
h2o
表示吸附相中h2o的浓度;y
h2o
表示原料汽中h2o的浓度;x
hdo
表示吸附相中hdo的浓度;y
hdo
表示原料汽中hdo的浓度;x
d2o
表示吸附相中d2o的浓度;y
d2o
表示原料汽中d2o的浓度。
[0100]
二、实验结果
[0101]
1、活化态fdc-1a和fdc-2a的晶体结构
[0102]
活化状态fdc-1a和fdc-2a的晶体结构如图6所示,其中,图6a为fdc-1a和fdc-2a的活化晶体结构图,图6b为fdc-1a和fdc-2a中的孔结构,图6c为fdc-1a的扩散通道和结构,图6d为fdc-2a的扩散通道和结构,从图6中可以看出,fdc-1a和fdc-2a是由许多相同尺寸的纳米孔笼和极窄的扩散通道组成,从一个孔笼到与之相邻的孔笼有6条相同的扩散通道。fdc-1a中所有扩散通道中都有一个的三角形“门”,这个“门”由两个间苯二甲酸和dbap中两个苯环围绕而成。同样,fdc-2a中的“门”是由两个间苯二甲酸和idb中两个苯环围绕而成,尺寸为略小于fdc-1a。由此可见,配体结构上的微调带来扩散通道的尺寸变化,这一变化虽然微小,但是对于分离具有非常小的分子动力学直径的水同位素体分子来说至关重要。
[0103]
2、活化态fdc-1a和fdc-2a的晶体稳定性
[0104]
将fdc-1a和fdc-2a分别在水(298k或363k)、甲醇(298k或333k)和n,n-二甲基甲酰胺(dmf)(298k或393k)中浸泡7天。图7为fdc-1a和fdc-2a在不同溶剂中浸泡7天后的pxrd谱图,其中,图7a为fdc-1a在不同溶剂中浸泡7天后的pxrd谱图,图7b为fdc-2a在不同溶剂中浸泡7天后的pxrd谱图,从图7中可以看出fdc-1a和fdc-2a在水、甲醇和dmf中浸泡后的pxrd曲线基本不变,说明fdc-1a和fdc-2a表现出优异的水热及溶剂稳定性。
[0105]
3、fdc-1a和fdc-2a吸附行为及其吸附过程动力学研究:
[0106]
首先研究了298k不同的相对压力(p/ps)下fdc-1a和fdc-2a对于h2o和d2o的吸-脱附曲线,结果如图8a和图8b所示,其中,横坐标代表压力,纵坐标代表吸附量,从图8a和图8b中可以看出,在298k的温度下,当p/ps为0.98时,fdc-1a和fdc-2a对h2o/d2o吸附率分别为1.39和1.64,说明fdc-1a和fdc-2a在水的同位素体里会优先吸附h2o。进一步研究了不同暴露时间下fdc-1a和fdc-2a对h2o和d2o的吸脱附曲线,如图8c和8d所示,从图中可以看出,随着暴露时间的延长,fdc-1a对h2o和d2o吸附量明显增加至约60ml g-1
,fdc-2a对h2o和d2o吸附量增加至约55ml g-1
,说明fdc-1a和fdc-2a的扩散动力学由扩散时间决定。图9为不同温度下本发明实施例中的配位聚合物吸附h2o和d2o的压力-扩散速率-吸附量全景图,其中x轴
为压力,y轴为扩散速率,z轴为吸附量,顺着箭头的方向,温度依次从278k增加到323k,其中,图9a为fdc-1a吸附h2o的压力-扩散速率-吸附量全景图,图9b为fdc-1a吸附d2o的压力-扩散速率-吸附量全景图,图9c为fdc-2a吸附h2o的压力-扩散速率-吸附量全景图,图9d为fdc-2a吸附d2o的压力-扩散速率-吸附量全景图,从图9中可以看出,当温度从278k增加至323k时,fdc-1a对h2o的吸附量从1.7ml g-1
显著增加到32.6ml g-1
,fdc-2a对h2o的吸附量从1.3ml g-1
增加到15.1ml g-1
,fdc-1a对d2o的吸附量从0.6ml g-1
增加到24.4ml g-1
,fdc-2a对d2o的吸附量从0.5ml g-1
增加到11.8ml g-1
。随着温度的升高,fdc-1a和fdc-2a对h2o和d2o的吸附量均明显增加,表明其吸附行为受温度控制。不同于常规多孔材料的气体/蒸汽吸附量是随温度升高而降低,该体系相反的吸附行为是典型的扩散受控体系的特征。
[0107]
4、不同温度下h2o和d2o扩散速率
[0108]
进一步研究了不同温度下h2o和d2o在本发明实施例中的配位聚合物中的扩散速率(水的同位素体扩散速率的测定方法是测量其在一定温度下的吸附曲线,记录每个测量点随时间变化的压力的原始数据,再通过crank方程求解,最终得到不同温度下水同位素体的扩散速率(ds))。不同温度下h2o和d2o的扩散速率结果如图9所示,从图9中可以看出,在低温下fdc-1a和fdc-2a对h2o或d2o的扩散速率都很小,随着温度和压力的升高,fdc-1a和fdc-2a对h2o或d2o的扩散速率逐渐增加。在298k时,fdc-1a和fdc-2a中h2o的扩散速率ds值分别为1.56
×
10-2
和1.11
×
10-2r2 s-1
,fdc-1a和fdc-2a中d2o的扩散速率ds值分别为2.05
×
10-3
和1.37
×
10-3r2 s-1
,明显看出在fdc-1a和fdc-2a中,h2o的扩散速率远远大于d2o,分别是d2o的7.6和8.1倍。另一方面,在298k时,fdc-1a中h2o和d2o的扩散速率是fdc-2a的1.4倍和1.5倍,表明fdc-1a的扩散动力学更快,fdc-2a比fdc-1a具有更小的扩散通道和更低的扩散速率。
[0109]
5、水同位素体分离的结果
[0110]
利用程序控温吸脱附(tpd)-质谱联用仪研究了在298k时fdc-1a和fdc-2a对水同位素体的混合蒸汽的动态分离性能。图10为298k温度下本发明实施例中的配位聚合物对水同位素体分离的麦凯布-蒂勒图,其中,横坐标为水同位素体原料蒸汽含量比例,纵坐标为吸附相中水同位素体吸附比例,图10a为fdc-1a对水同位素体分离的麦凯布-蒂勒图,图10b为fdc-2a对水同位素体分离的麦凯布-蒂勒图,从图10a和图10b可以明显看出在h2o/hdo/d2o的三元共混蒸汽中,fdc-1a和fdc-2a在0.5h的吹扫过程中,选择性的从h2o/hdo/d2o混合物中吸附h2o,产生明显的h2o富集。即使在h2o:hdo:d2o=0.3:9.3:90.4的混合蒸汽中,fdc-1a和fdc-2a的h2o吸附比例仍然分别达到26.8%和38.6%,其对应的h2o分离因子分别为124和212,如图11所示,图11为298k温度下本发明实施例中的配位聚合物吸附相中h2o分离因子随原料蒸汽中h2o含量变化的情况,其中,横坐标为h2o在水同位素体原料中的比例,纵坐标为h2o的分离因子。从图中可以看出,fdc-2a对h2o的吸附比例和h2o分离因子高于fdc-1a,这归因于fdc-2a对水同位素体有更严格的扩散控制,进一步放大了水同位素体扩散速率的微小差异。
[0111]
通过上述研究可以表明,本发明实施例中的配位聚合物之所以能够在278~473k的范围内对水的同位素体进行分离是由于本发明实施例中的配位聚合物可以允许水同位素体通过并控制其扩散,能够放大水同位素体之间扩散速率的细微差异,实现对h2o的高选择性吸附和分离。
[0112]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的
限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1