一种手性苯乙醇的连续制备方法与流程
1.本发明涉及手性苯乙醇合成领域,特别涉及一种手性苯乙醇的连续制备方法。
背景技术:
2.目前,手性芳香醇是许多医药、农药、精细化工和香料的中间体,也是卤化物、酯类和氨基化合物等多种有机化合物的前体,通过手性芳香醇的光学特性可以将手性中心在复杂有机物中得到延伸,从而得到具有光学活性的手性化合物。
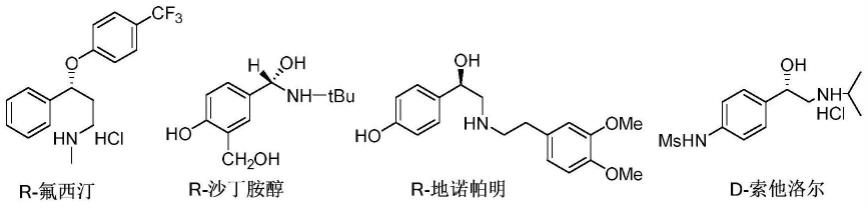
3.将其不对称合成方法从实验室研究走向工业化应用是一件急需研究的课题,以来满足人们对手性化学品日益增长的需求,特别是手性药物领域的需求。而连续流技术因更安全有效,传质传热效率更强,环保节约,符合绿色化学发展的方向。如图1所示,为四种含有手性芳香醇结构的药物分子。
4.目前,常见的手性苯乙醇合成方法为:以苯乙酮为原料,使之在高压反应釜中反应,生成手性苯乙醇。但是该种合成方法中对催化剂以及底物浓度的要求很高,限制了工业化生产。因此,亟需一种更安全有效、性质稳定、传质传热效率更强的方法。
技术实现要素:
5.针对现有技术的不足,本发明提出了一种手性苯乙醇的连续制备方法,该合成方法高效、安全性高,主要通过使用连续流反应,连续合成手性苯乙醇。本技术中手性苯乙醇的转化率达68%以上,对映体过量值达到94.5%以上。因此,本技术制备的手性苯乙醇具有产品品质高以及收率较高的优点。
6.为解决上述技术问题,本发明提供一种手性苯乙醇的连续制备方法,其特征在于,至少包括以下步骤:
7.s1、制备络合催化剂;
8.s2、取有机溶剂、叔丁醇、苯乙酮、助催化剂,混合溶解过滤后,滤液为用于连续流反应的反应液;
9.s3、连续反应装置加压至8-20atm,温度控制在25-30℃,反应液以0.5-1ml/min、氢气流量以20-30sccm,通入连续反应装置中,当反应达到稳态后开始收集粗产物,粗产物提纯后得到手性苯乙醇。
10.进一步地,所述苯乙酮与所述络合催化剂的物质的量比为(1000-3000):1。
11.进一步地,步骤s1为,二氯苯基钌(ii)二聚体与(s,s)-diop生成暗红色固体,暗红色固体与咪唑配体生成络合催化剂;反应均在有机溶剂中进行。
12.进一步地,所述二氯苯基钌(ii)二聚体与(s,s)-diop的物质的量比为1:(1.8-2.2)。
13.进一步地,所述咪唑配体为(s)-1-(1h-苯并[d]咪唑-2-基)乙烷-1-胺,所述暗红色固体与咪唑配体的质量比为8:(2-3)。
[0014]
进一步地,所述苯乙酮与所述助催化剂的质量比为(16-20):1。
[0015]
进一步地,连续流反应前需要彻底排空连续反应装置内部的空气以及其他杂质,具体步骤为,在氮气流量为20-30sccm条件下,脱气无水甲苯以4-6ml/min冲洗反应装置20-30min。
[0016]
进一步地,所述助催化剂为氢氧化钾、氢氧化钠、甲醇钠、乙醇钠、氢化钠、叔丁醇钠、叔丁醇钾中的任意一种。
[0017]
进一步地,所述有机溶剂为n,n-二甲基甲酰胺、甲苯、二氯甲烷、氯仿中的任意一种。
[0018]
进一步地,所述手性苯乙醇的转化率达68%以上,对映体过量值达到94.5%以上。
[0019]
综上所述,本发明具有以下有益效果:
[0020]
1、本技术为连续流反应器在手性苯乙醇合成中的应用,本发明具有方式新颖,操作简单实用,产品品质及收率较高的优点,从而使生产成本降低,同时对环境友好污染较少。
[0021]
2、本技术中得到的产物分离处理方便,且工艺流程操作简单,反应条件相对温和,且污染相对较小。最后通过本发明工艺流程可以有效地避免现有的反应无法较大规模生产的缺点,更加有利于实现工业上的大规模生产要求,同时提高了手性苯乙醇的品质以及收率。
[0022]
3、本技术采用连续流合成方法,强化了反应的传质、传热性能,保持反应温度恒定,避免了反应时出现飞温,冲料,反应失控等现象,可操作性和安全性大大提高。
附图说明
[0023]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图示出的结构获得其他的附图。
[0024]
图1为背景技术中四种含有手性芳香醇结构的药物分子。
[0025]
图2为实施例1中(s)-苯乙醇的核磁氢谱图。
[0026]
图3为连续流反应流程图。
[0027]
其中,1、进料口;2、泵;3、第一安全阀;4、废液排出口;5、第一压力表;6、第二压力表;7、第二安全阀;8、第一废气排出口;9、氢气罐;10、反应线圈;11、温控装置;12、气液分离器;13、出料口;14、第二废气排出口;15、氮气罐;16、氢气阀门;17、氮气阀门;18、出料口阀门。
[0028]
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
[0029]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0030]
下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试
验材料和试剂等,如无特殊说明,均可从商业途径获得。以下实施例中的定量试验,均设置三次重复实验,数据为三次重复实验的平均值或平均值
±
标准差。
[0031]
另外,全文中的“和/或”包括三个方案,以a和/或b为例,包括a技术方案、b技术方案,以及a和b同时满足的技术方案;另外,各个实施例之间的技术方案可以相互结合,但是必须是以本领域普通技术人员能够实现为基础,当技术方案的结合出现相互矛盾或无法实现应当认为这种技术方案的结合不存在,也不在本发明要求的保护范围之内。
[0032]
实施例1
[0033]
制备络合催化剂:
[0034]
精确称取16.3mg二氯苯基钌(ii)二聚体和32.5mg(s,s)-diop(32mm),加入2ml n,n-二甲基甲酰胺中,得到第一混合溶液,第一混合溶液在100℃下搅拌30min。反应完成后,在高真空条件下去除有机溶剂,得到暗红色固体;
[0035]
精确称取上述生成的8mg暗红色固体和3mg咪唑配体,置于鸡心瓶中,加入10ml二氯甲烷,得到第二混合溶液,将氩气通过第二混合溶液3min,使之脱气;室温搅拌5min后,在真空条件下除去溶剂,得到络合催化剂,其中咪唑配体为(s)-1-(1h-苯并[d]咪唑-2-基)乙烷-1-胺。
[0036]
上述络合催化剂的制备在手套箱中完成。
[0037]
配置反应液:
[0038]
分别取36ml脱气无水甲苯、4ml叔丁醇和1.2ml苯乙酮(s/c=1000),加入67mg叔丁醇钾以及0.01mmol络合催化剂,混合溶解后得到第三混合溶液,将第三混合溶液过滤,滤液加入鸡心瓶中,滤液为用于连续流反应的反应液。
[0039]
连续流反应:
[0040]
如图3所示,脱气无水甲苯(4ml/min)从进料口1通入,同时开启氢气罐9位置的氢气阀门16,保持氢气的流量为20sccm。氢气以及脱气无水甲苯冲洗反应装置20min后,保证彻底排空反应装置内部的空气以及其他杂质。冲洗后的废液经过气液分离器12后,从出料口13排出;废气从第二废气排出口14排出。
[0041]
当反应装置内部的杂质除尽后,将反应装置加压至8atm,使用温控装置11将反应线圈10的温度控制在25℃,此时,上述配置完成的反应液通过泵2输送至反应线圈10,反应液的流速为1ml/min;并且同步输送氢气,氢气的流量保持在20sccm。在反应过程中,如果检测到第一压力表5和第二压力表6显示的压力值过大,达到第一安全阀3和第二安全阀7设定的压力后,打开第一安全阀3和第二安全阀7,使反应体系中多余的液体和气体从废液排出口4和第一废气排出口8排出,反应体系压力瞬间降低。
[0042]
当反应达到稳态后,粗产物聚集于气液分离器12中,开启出料口阀门13,收集出料口13处的粗产物,收集好的粗产物通过硅胶柱(石油醚/乙酸乙酯=10:1)分离提纯得到产物(s)-苯乙醇,(s)-苯乙醇的转化率和ee值分别为68%和99.3%。
[0043]
同时,产物(s)-苯乙醇的核磁共振氢谱图如图2所示,图2可以证明产物(s)-苯乙醇的生成。
[0044]
反应结束后,缓慢开启氮气罐15处的氮气阀门17,通过氮气调控反应体系压力;之后于出料口13位置通入无水甲苯清洗反应装置10min,再通入脱气无水乙醇冲洗10min。
[0045]
实施例2-6与实施例1的不同之处在于,连续流反应的反应条件参数不同。
[0046]
实施例2-6中连续流反应条件参数如表1所示。
[0047]
表1各实施例中连续流反应条件参数
[0048] 实施例1实施例2实施例3实施例4实施例5实施例6压力/atm8888820温度/℃252525302530反应液流速/ml
·
min-1
10.50.50.50.50.5氢气流量/sccm202030354040
[0049]
实施例1至实施例6中制备得到的(s)-苯乙醇的转化率以及ee值如表2所示。
[0050]
表2实施例1至实施例6中制备得到的(s)-苯乙醇的转化率以及ee值
[0051] 转化率/%ee/%实施例16899.3实施例27299.5实施例39994.7实施例49996.3实施例59997.5实施例69994.5
[0052]
对比例1与实施例2的区别在于,反应过程中,连续反应装置的温度为45℃,其产物的转化率和ee值分别为99%和65.5%。由此可以看出,反应温度上升,反应的转化率上升,但是,相对应的(s)-苯乙醇的含量下降。
[0053]
对比例2与实施例6的区别在于,催化剂的用量降低一倍。其产物的转化率和ee值分别为68%和49%。由此可以看出,催化剂的含量对反应的转化率和ee选择性均有比较重要的影响。
[0054]
对比例3与实施例2的区别在于,反应过程中,连续反应装置的压力为30atm,其产物的转化率和ee值分别为96%和93.2%。
[0055]
对比例4与实施例2的区别在于,反应过程中,反应液的流速为2ml/min,其产物的转化率和ee值分别为59.6%和97%。
[0056]
对比例5与实施例2的区别在于,反应过程中,氢气的流量为40sccm,其产物的转化率和ee值分别为99%和95.7%。
[0057]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载范围。
[0058]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1