一种淫羊藿素的制备方法与流程
1.本发明涉及医药领域,具体涉及一种淫羊藿素的制备方法。
背景技术:
2.淫羊藿是我国的一种传统中草药,具有“补肾阳、强筋骨和祛风湿”的功效,2015版《中华人民共和国药典》中共记载了5种淫羊藿,分别为淫羊藿、柔毛淫羊藿、箭叶淫羊藿、朝鲜淫羊藿和巫山淫羊藿。上述5种淫羊藿中含有大量的淫羊藿总黄酮,包括淫羊藿素、淫羊藿苷和朝藿定a~c等。有研究表明,淫羊藿素具有优异的抗肿瘤效果,能够对肝癌、前列腺癌和胃癌等均有不同程度的抑制作用,尤其对肝癌具有很好的治疗效果,可以明显抑制肝癌的细胞恶性生长。但淫羊藿、柔毛淫羊藿、箭叶淫羊藿和朝鲜淫羊藿中的质量成分为淫羊藿苷(含量大于0.50%),巫山淫羊藿的质量控制成分为朝藿定c(含量>1%),即淫羊藿中存在的淫羊藿素的量较少。
3.而淫羊藿素可以通过淫羊藿苷和朝藿定c转化得到,因此将淫羊藿苷和朝藿定c转化为淫羊藿素的方法受到众多学者的广泛关注。现有技术中,多采用酸解、酶解或酸解-酶解联用的方式将淫羊藿苷和朝藿定c转化为淫羊藿素,但酸解大多采用盐酸或硫酸等,导致最终得到的淫羊藿素中有cl-和so
42-的残留,用于制备肝癌药物,会导致其在人体内残留,不利于人体安全。另外,以上述淫羊藿药材为原料制备得到的淫羊藿素的得率还有待提升。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种淫羊藿素的制备方法。所述制备方法采用有机酸(柠檬酸和/或草酸)进行酸解,均为可食用酸,人体摄入后不会造成危害,且通过有机酸酸解-酶解联用的方式使得从淫羊藿药材中制备淫羊藿素的得率大幅提高。
5.为达到此目的,本发明采用以下技术方案:
6.第一方面,本发明提供一种淫羊藿素的制备方法,包括以下步骤:
7.(1)将淫羊藿提取物与有机酸混合进行酸解,得到酸解产物;
8.其中,所述淫羊藿提取物包括淫羊藿苷和/或朝藿定c;
9.所述有机酸包括柠檬酸和/或草酸;
10.(2)将酸解产物与酶混合进行酶解,得到淫羊藿素。
11.优选地,所述有机酸为草酸。
12.优选地,所述酶包括纤维素酶、β-葡萄糖苷酶、蜗牛酶或柚柑酶中的任意一种或多种,更优选为纤维素酶。
13.优选地,所述酸解中淫羊藿提取物与有机酸的摩尔比为1:(100~300)。
14.进一步优选地,所述酸解中淫羊藿提取物与有机酸的摩尔比为1:(200~300)。
15.优选地,所述酸解的温度为50~65℃,所述酸解的时间为24~48h。
16.进一步优选地,所述酸解的温度为55~65℃,所述酸解的时间为36~48h。
17.优选地,所述淫羊藿提取物与缓冲溶液的质量体积比为1:(2~4)mg/ml。
18.优选地,所述淫羊藿提取物与酶的质量比为1:(1.5~3)。
19.优选地,所述酶解的温度为45~55℃,所述酶解的时间为20~36h。
20.与现有技术相比,本发明的有益效果为:
21.本发明以有机酸酸解-酶解联用的方式将淫羊藿药材和巫山淫羊藿药材中的淫羊藿苷和/或朝藿定c转化为淫羊藿素,制备得到的淫羊藿素得率高,其中淫羊藿药材中淫羊藿素的得率可达到5.116%,提高了44.1倍,巫山淫羊藿药材中淫羊藿素的得率可达到1.441%,提高了35.2倍,且酸解过程中使用的有机酸均为可食用酸,对人体没有危害。
附图说明
22.图1为朝藿定c的标准曲线图;
23.图2为淫羊藿苷的标准曲线图;
24.图3为淫羊藿素的标准曲线图;
25.图4为淫羊藿素在点板过程中的展开结果图;
26.图5为淫羊藿苷和朝藿定c在点板过程中的展开结果(从左至右依次为淫羊藿苷和朝藿定c;
27.图6为实施例3中在酸解48h时不同体系的点板结果图;
28.其中,
①
淫羊藿苷+柠檬酸,
②
淫羊藿苷+草酸,
③
淫羊藿苷+醋酸,
④
朝藿定c+柠檬酸,
⑤
朝藿定c+草酸,
⑥
朝藿定c+醋酸;
29.图7为淫羊藿苷标准品的液相图;
30.图8为朝藿定c的标准品的液相图;
31.图9为淫羊藿苷+1:100摩尔比草酸体系反应24h后得到的产物液相图;
32.图10为淫羊藿苷+1:200摩尔比草酸体系反应24h后得到的产物液相图;
33.图11为淫羊藿苷+1:300摩尔比草酸体系反应24h后得到的产物液相图;
34.图12为淫羊藿苷+1:100摩尔比草酸体系反应48h后得到的产物液相图;
35.图13为淫羊藿苷+1:200摩尔比草酸体系反应48h后得到的产物液相图;
36.图14为淫羊藿苷+1:300摩尔比草酸体系反应48h后得到的产物液相图;
37.图15为朝藿定c+1:100摩尔比草酸体系反应24h后得到的产物液相图;
38.图16为朝藿定c+1:200摩尔比草酸体系反应24h后得到的产物液相图;
39.图17为朝藿定c+1:300摩尔比草酸体系反应24h后得到的产物液相图;
40.图18为朝藿定c+1:100摩尔比草酸体系反应48h后得到的产物液相图;
41.图19为朝藿定c+1:200摩尔比草酸体系反应48h后得到的产物液相图;
42.图20为朝藿定c+1:300摩尔比草酸体系反应48h后得到的产物液相图;
43.图21为淫羊藿苷+1:200摩尔比草酸体系反应48h后得到的产物液质图(正离子模式);
44.图22为淫羊藿苷+1:200摩尔比草酸体系反应48h后得到的产物液质图(负离子模式);
45.图23为朝藿定c+1:200摩尔比草酸体系反应48h后得到的产物液质图(正离子模式);
46.图24为朝藿定c+1:200摩尔比草酸体系反应48h后得到的产物液质图(负离子模
式);
47.图25为实施例5中淫羊藿苷酸水解+葡聚糖酶酶解体系得到的产物液相图;
48.图26为实施例5中朝藿定c酸水解+葡聚糖酶酶解体系得到的产物液相图;
49.图27为实施例5中淫羊藿苷酸水解+纤维素酶酶解体系得到的产物液相图;
50.图28为实施例5中朝藿定c酸水解+纤维素酶酶解体系得到的产物液相图;
51.图29为实施例6中淫羊藿苷酸水解+纤维素酶酶解体系得到的产物液相图;
52.图30为实施例6中朝藿定c酸水解+纤维素酶酶解体系得到的产物液相图;
53.图31为实施例7中淫羊藿药材在醇-水体系中提取产物的液相图;
54.图32为实施例7中淫羊藿药材在醇-水体系+纤维素酶酶解体系中提取产物的液相图;
55.图33为实施例7中淫羊藿药材在醇-酸体系中提取产物的液相图;
56.图34为实施例7中淫羊藿药材在醇-酸体系+纤维素酶酶解体系中提取产物的液相图;
57.图35为实施例7中巫山淫羊藿药材在醇-水体系中提取产物的液相图;
58.图36为实施例7中巫山淫羊藿药材在醇-水体系+纤维素酶酶解体系中提取产物的液相图;
59.图37为实施例7中巫山淫羊藿药材在醇-酸体系中提取产物的液相图;
60.图38为实施例7中巫山淫羊藿药材在醇-酸体系+纤维素酶酶解体系中提取产物的液相图。
具体实施方式
61.下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
62.本发明所涉及的所有原料,对其来源没有特别的限制,从市场上购买或者按照本领域技术人员熟知的常规技术手段制备得到即可。
63.本发明提供一种淫羊藿素的制备方法,包括以下步骤:
64.(1)将淫羊藿提取物与有机酸混合进行酸解,得到酸解产物;
65.其中,所述淫羊藿提取物包括淫羊藿苷和/或朝藿定c;
66.所述有机酸包括柠檬酸和/或草酸;
67.(2)将酸解产物与酶混合进行酶解,得到淫羊藿素。
68.按照本发明,首先将淫羊藿提取物与有机酸混合进行酸解,得到酸解产物。所述淫羊藿提取物优选包括淫羊藿苷和/或朝藿定c,可以从市场上购买或者从淫羊藿药材或巫山淫羊藿药材中进行天然提取而得到。所述提取的溶剂包括水、乙醇水溶液或无水乙醇中的任意一种或多种。本发明对提取的方式没有特别的限制,按照本领域技术人员熟知的常规技术手段即可。所述有机酸优选包括柠檬酸和/或草酸,更优选为草酸。在本发明中,所述淫羊藿提取物与有机酸的摩尔比优选为1:(100~300),所述酸解的温度为50~65℃,所述酸解的时间优选为24~48h。经研究发现,在酸解过程中,淫羊藿苷和朝藿定c的酸水解产物优
先生成淫羊藿素7位连接一分子葡萄糖,继而生成脱水淫羊藿素7位连接一分子葡萄糖,但脱水淫羊藿素7位连接一分子葡萄糖不稳定,在加大酸用量以及酸解时间的条件下,脱水淫羊藿素7位连接一分子葡萄糖可以通过水合作用生成淫羊藿素7位连接一分子葡萄糖,即酸解的目标产物。因此,在本发明中,所述淫羊藿提取物与有机酸的摩尔比更优选为1:(200~300),最优选为1:300,所述酸解的时间更优选为36~48h,最优选为48h。
69.按照本发明,在得到酸解产物后,对酸解产物进行酶解。为了保证酶解过程中酶的反应活性,在得到酸解产物后,优选将酸解产物与缓冲溶液混合,将体系的ph调至5~7,所述缓冲溶液优选为醋酸/醋酸钠缓冲液,所述淫羊藿提取物与缓冲溶液的质量体积比优选为1:(2~4)mg/ml,更优选为1:(2~3)mg/ml。在本发明中,所述酶解过程中使用的酶优选包括纤维素酶、β-葡萄糖苷酶、蜗牛酶或柚柑酶中的任意一种或多种,从成本角度考虑,更优选为纤维素酶。将纤维素酶作用于酸解产物,经液相检测结果可知,能够得到淫羊藿素。在本发明中,所述淫羊藿提取物与酶的质量比优选为1:(1.5~3),更优选为1:(2~3)。所述酶解的温度优选为45~55℃,更优选为50~55℃,所述酶解的时间优选为20~36h,更优选为24~36h。
70.需要说明的是,在淫羊藿素的整个制备过程中,本发明采用点板监测反应进展,并通过标准曲线确定最终淫羊藿素的得率。
71.在本发明中,所述点板时,朝藿定c和淫羊藿苷展开的流动相优选为三氯甲烷:甲醇:水:冰乙酸=7:3:1:0.3(体积比),所述淫羊藿素展开的流动相优选为二氯甲烷:乙酸乙酯:冰乙酸=8:1:0.3(体积比),以得到各组分合适的rf值,在监测过程中对体系内的组分进行定性分析。
72.在本发明中,所述标准曲线的绘制过程没有特别的限制,按照本领域技术人员熟知的常规技术手段即可。色谱条件优选如下:以十八烷基硅烷键合硅胶为填充剂,以乙腈(a)-水(b)为流动相进行梯度洗脱,洗脱条件:0-30min,25%a;30-42min,25%-45%a;42-55min,45%a;55-65min,45%-75%a;65-77min,75%a;77-90min,75%-25%a。流速0.8ml/min,检测波长为270mn,柱温30℃,进样量20μl。
73.本发明所提供的淫羊藿素的制备方法是为了满足实际应用需要的,因此本发明参考上述方法,以淫羊藿药材和巫山淫羊藿药材为原料,提取淫羊藿素。本发明优选将淫羊藿药材或巫山淫羊藿药材与醇-酸混合溶液进行酸提,直接一步得到酸提产物。本发明对酸提的方式没有特别的限制,按照本领域技术人员熟知的常规技术手段即可,本发明优选在70~90℃下回流提取2~4h,更优选在80~90℃下回流提取3~4h。所述醇-酸混合溶液中,醇优选包括50vol%乙醇水溶液和/或无水乙醇,更优选为无水乙醇,所述酸优选包括柠檬酸和/或草酸,更优选为草酸,最优选为草酸的饱和溶液,所述酸与醇的体积比优选为1:(1~2),更优选为1:1。为了提高淫羊藿素的得率,在酸提结束后,优选将酸提液抽滤得到第一次滤液,将滤渣按照上述方式再进行重复酸提,所述重复的次数不低于1次,将后续抽滤得到的滤液与第一次滤液合并,得到合并滤液。
74.按照本发明,酸提结束后进行酶解,优选将合并滤液调至ph为6~7,滤去析出物后,将合并滤液浓干再加入甲醇溶解,再滤去不溶性物质,再浓干后,进行后续的酶解处理。本发明优选在最终得到的浓干产物中,加入缓冲溶液后,再加入酶进行酶解。所述缓冲溶液优选为ph在5~6的醋酸/醋酸钠缓冲液,所述酶优选包括纤维素酶、β-葡萄糖苷酶、蜗牛酶
或柚柑酶中的任意一种或多种,更优选为纤维素酶,所述酶解的温度优选为45~55℃,更优选为50~55℃,所述酶解的时间优选为20~36h,更优选为24~36h。
75.按照本发明,所述酶解结束后,优选对酶解产物进行后处理,所述后处理包括将酶解产物浓干后,加入甲醇溶解,滤去不溶性物质,得到滤液,滤液中含有目标产物-淫羊藿素。
76.为了进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例中所用的实验原料没有特殊的限制,均可以从市场上购买或者按照本领域技术人员熟知的常规技术手段得到。
77.实施例1
78.本实施例用于绘制朝藿定c、淫羊藿苷和淫羊藿素的标准曲线,步骤如下:
79.精密称取朝藿定c 25.3mg、淫羊藿苷25.4mg、淫羊藿素25.2mg,加入甲醇溶解,于50ml容量瓶中定容,配制成浓度为0.5mg/ml的混合对照品溶液。精密量取对照品溶液,分别稀释2、4、8、16、32、64、128、256倍,得各浓度梯度的溶液,取上述各溶液分别进样20μl,记录峰面积。以峰面积积分值(y)为纵坐标、对照品的浓度(x)为横坐标,绘制标准曲线。
80.色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈(a)-水(b)为流动相进行梯度洗脱,洗脱条件:0-30min,25%a;30-42min,25%-45%a;42-55min,45%a;55-65min,45%-75%a;65-77min,75%a;77-90min,75%-25%a。流速0.8ml/min,检测波长为270mn,柱温30℃,进样量20μl。
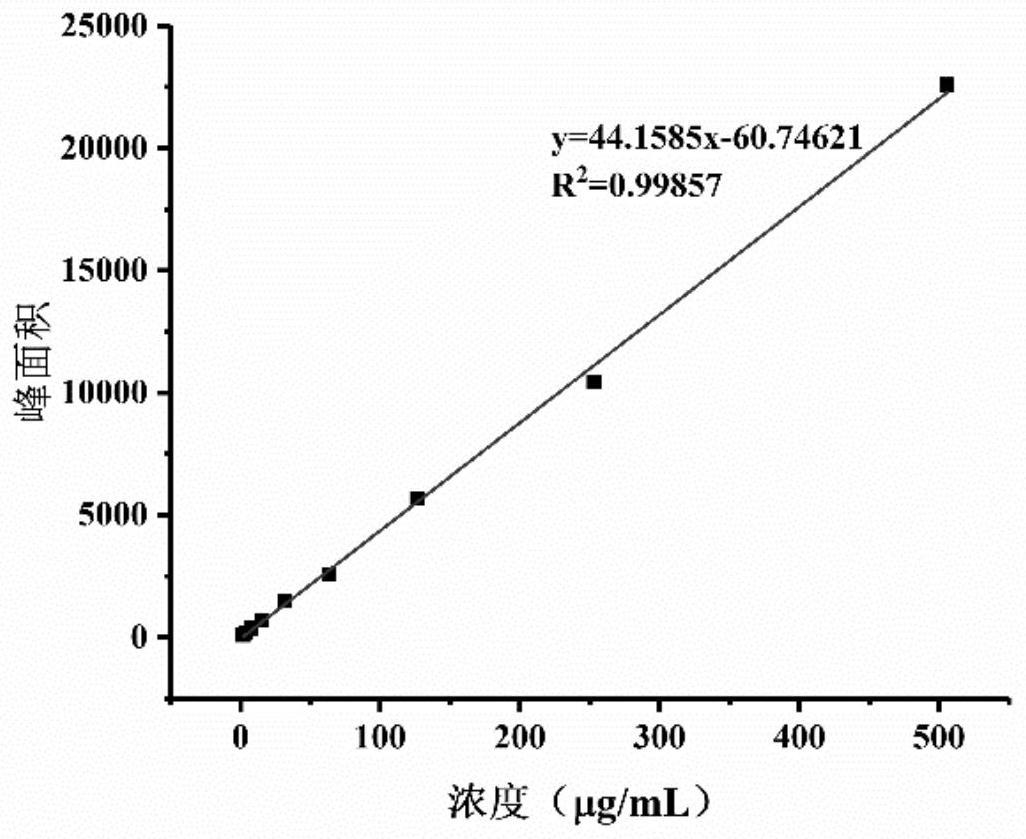
81.其中,朝藿定c的标准曲线如图1所示:y=44.1585x-60.74261,r2=0.99857;
82.淫羊藿苷的标准曲线如图2所示:y=53.37934x-61.15418,r2=0.99897;
83.淫羊藿素的标准曲线如图3所示:y=81.24921x+226.36158,r2=0.99905。
84.实施例2
85.本实施例对朝藿定c、淫羊藿苷和淫羊藿素进行薄层层析,步骤如下:
86.称取朝藿定c 5mg、淫羊藿苷5mg、淫羊藿素5mg,加入5ml甲醇溶解,用于点板。结合点板结果和药典,朝藿定c和淫羊藿苷展开的流动相为三氯甲烷:甲醇:水:冰乙酸=7:3:1:0.3,淫羊藿素展开的流动相为二氯甲烷:乙酸乙酯:冰乙酸=8:1:0.3;
87.其中,淫羊藿素的展开结果如图4所示,rf(淫羊藿素)=2.3/3.4=0.676;
88.淫羊藿苷和朝藿定c的展开结果如图5所示(从左至右依次为淫羊藿苷和朝藿定c),rf(淫羊藿苷)=2.3/3.5=0.686,rf(朝藿定c)=1.7/3.5=0.486。
89.根据上述的薄层条件,在后续的实施例中用于监测反应进展。
90.实施例3
91.本实施例用于确定有机酸水解条件,步骤如下:
92.精密称取朝藿定c、淫羊藿苷各10mg,加入10ml甲醇分别溶解,得储备液。各取朝藿定c和淫羊藿苷的储备液三份,每份1ml,以1:100摩尔比分别加入草酸、柠檬酸和醋酸溶液各1ml,于60℃下水浴中进行水解,分别于12h、24h、36h、48h取样点板,监测反应进展。
93.取48h的点板结果,如图6所示,图中从左至右,依次为:
①
淫羊藿苷+柠檬酸、
②
淫羊藿苷+草酸、
③
淫羊藿苷+醋酸、
④
朝藿定c+柠檬酸、
⑤
朝藿定c+草酸、
⑥
朝藿定c+醋酸。由图6可知,朝藿定c在三种酸作用下,点板均未见新点出现,表明其在有机酸条件下未水解;淫羊藿苷在柠檬酸和草酸作用下点板均有新点出现,且草酸作用强于柠檬酸,醋酸作用最
弱,在醋酸作用下未有新点出现,根据以上结果,选用草酸进行后续酸水解。
94.实施例4
95.本实施例用于确定草酸水解条件,步骤如下:
96.参考实施例3,制备朝藿定c和淫羊藿苷的储备液,取两种储备液各3份,每份1ml,分别以1:100、1:200、1:300摩尔比加入草酸溶液1ml,于60℃下进行水浴,分别于24h、48h取样,液相分析(色谱条件:以十八烷基硅烷键合硅胶为填充剂,以乙腈(a)-水(b)为流动相进行梯度洗脱,洗脱条件:0-30min,25%a;30-42min,25%-45%a;42-55min,45%a;55-65min,45%-75%a;65-77min,75%a;77-90min,75%-25%a。流速0.8ml/min,检测波长为270mn,柱温30℃,进样量20μl),监测反应进行情况。
97.另外,取朝藿定c和淫羊藿苷的储备液,按1:200摩尔比加入草酸溶液1ml,反应48h后的液体进行液质分析(离子源esi,正负离子扫描模式均用;离子源电压3kv;离子源温度250℃;质谱接口温度300℃;扫描范围50-1000m/z;扫描方式多反应监测mrm),作为液相分析结果的参比。
98.淫羊藿苷的标准品的液相图如图7所示,朝藿定c的标准品的液相图如图8所示。淫羊藿苷+1:100摩尔比草酸+反应24h的液相图如图9所示,淫羊藿苷+1:200摩尔比草酸+反应24h的液相图如图10所示,淫羊藿苷+1:300摩尔比草酸+反应24h的液相图如图11所示,淫羊藿苷+1:100摩尔比草酸+反应48h的液相图如图12所示,淫羊藿苷+1:200摩尔比草酸+反应48h的液相图如图13所示,淫羊藿苷+1:300摩尔比草酸+反应48h的液相图如图14所示,朝藿定c+1:100摩尔比草酸+反应24h的液相图如图15所示,朝藿定c+1:200摩尔比草酸+反应24h的液相图如图16所示,朝藿定c+1:300摩尔比草酸+反应24h的液相图如图17所示,朝藿定c+1:100摩尔比草酸+反应48h的液相图如图18所示,朝藿定c+1:200摩尔比草酸+反应48h的液相图如图19所示,朝藿定c+1:300摩尔比草酸+反应48h的液相图如图20所示,淫羊藿苷+1:200摩尔比草酸+反应48h的液质图如图21~22所示,朝藿定c+1:200摩尔比草酸+反应48h的液质图如图23~24所示。
99.根据液质分析结果,结合文献(杨轶舜,张彤,丁越,陈嘉雯,周奕嘉,赵日吉,吴华燕,宋泽家.3种淫羊藿苷次生产物的制备及hplc测定[j].中成药,2020,42(03):779-782)和专利cn200710179673.8、cn200680044755.5进行分析,两标准品的酸水解产物在44min处出峰,解析确定为脱水淫羊藿素7位连接一分子葡萄糖(产物1),在38min处出峰,解析确定为淫羊藿素7位连接一分子葡萄糖(产物2);结合液相结果和两底物的结构可知,酸水解过程首先生成产物2,继而产物1在酸的作用下水合得到产物1,且酸的用量越大、反应时间越长,产物1向产物2转化的趋势越明显。因此,拟采用48h、1:300摩尔的酸水解条件作为后续实验条件。
[0100]
实施例5
[0101]
本实施例用于在草酸水解的前提下确定酶解条件,步骤如下:
[0102]
精密称取朝藿定c和淫羊藿素各10mg,配制成2mg/ml的甲醇溶液,各取两份溶液,每份1ml,均加入摩尔比1:300的草酸溶液1ml,于60℃下反应48h,反应结束后调ph至5-6,加入5ml 0.5m的醋酸/醋酸钠缓冲液,然后在各两份相同反应液中,一份加入葡聚糖酶4mg,一份加入纤维素酶4mg,于50℃下反应24h,取样,进行液相检测。
[0103]
淫羊藿苷酸水解+葡聚糖酶酶解的液相图如图25所示,朝藿定c酸水解+葡聚糖酶
酶解的液相图如图26所示,淫羊藿苷酸水解+纤维素酶酶解的液相图如图27所示,朝藿定c酸水解+纤维素酶酶解的液相图如图28所示。
[0104]
结合图25~28可知,葡聚糖酶作用于酸水解产物,液相检测结果显示并未在淫羊藿素出峰位置(65min左右)出峰,表明其并不能酶解酸水解产物得到淫羊藿素。纤维素酶作用于酸水解产物,液相检测结果显示在淫羊藿素出峰位置(65min左右)出峰,表明其能酶解酸水解产物得到淫羊藿素,且朝藿定c转化的淫羊藿素产率为9.9%,淫羊藿苷转化的淫羊藿素产率为14.9%。但是淫羊藿苷反应产物在74min处有一个很强的峰,推测其为淫羊藿苷的苷元,表明苷元向淫羊藿素转化得不完全,需加大酸的用量。
[0105]
实施例6
[0106]
本实施例加大酸的用量,计算淫羊藿素的得率,步骤如下:
[0107]
取实施例5配制的2mg/ml朝藿定c和淫羊藿苷的甲醇溶液各1ml,分别加入2ml草酸饱和溶液,于60℃下反应48h,反应结束后调ph至5-6,加入5ml 0.5m的醋酸/醋酸钠缓冲液,然后再分别加入纤维素酶4mg,于50℃下反应24h,反应结束后取样进液相检测。
[0108]
淫羊藿苷酸水解+纤维素酶酶解的液相图如图29所示,朝藿定c酸水解+纤维素酶酶解的液相图如图30所示。
[0109]
经计算,朝藿定c转化的淫羊藿素产率为15.72%,淫羊藿苷转化的淫羊藿素产率为18.54%,可以看出加大酸的用量使得朝藿定c和淫羊藿苷转化得到淫羊藿素的产率提高。
[0110]
实施例7
[0111]
本实施例以淫羊藿和巫山淫羊藿为原料,制备淫羊藿素,步骤如下:
[0112]
使用粉碎机将淫羊藿药材和巫山淫羊藿药材打成粉末,分别称取淫羊藿药材和巫山淫羊藿药材5.00g,各两份,置于4个250ml圆底烧瓶中,其中一份加入100ml 50vol%乙醇溶液,另一份加入50ml无水乙醇与50ml草酸的饱和溶液,于80℃油浴下,回流提取4h。提取结束后,将提取液抽滤得滤液1,滤渣再经上述方式提取一次,抽滤提取液得滤液2,将各两次提取液合并,并调ph至6-7,然后滤去析出物,使用旋蒸将滤液浓干。之后,加入甲醇50ml,超声溶解,滤去不溶性物质,取各滤液少许,液相检测。而后,再次使用旋蒸将滤液浓干,向浓干产物中加入100ml ph=5的醋酸/醋酸钠溶液,超声分散,再加入纤维素酶200mg,于50℃油浴下反应24h。反应结束,使用旋蒸将反应液浓干,浓干后加入甲醇50ml,超声溶解,滤去不溶性物质,取各滤液少许,再次液相检测。
[0113]
其中,淫羊藿(醇-水体系)提取结果的液相图如图31所示,淫羊藿(醇-水体系+纤维素酶酶解)的液相图如图32所示,淫羊藿(醇-酸体系)提取结果的液相图如图33所示,淫羊藿(醇-酸体系+纤维素酶酶解)的液相图如图34所示。巫山淫羊藿(醇-水体系)提取结果的液相图如图35所示,巫山淫羊藿(醇-水体系+纤维素酶酶解)的液相图如图36所示,巫山淫羊藿(醇-酸体系)提取结果的液相图如图37所示,巫山淫羊藿(醇-酸体系+纤维素酶酶解)的液相图如图38所示。
[0114]
由图31~34可知,淫羊藿(醇-水体系)无法提取到淫羊藿素;淫羊藿(醇-水体系+纤维素酶酶解)可以提取到淫羊藿素,其峰面积为1172.1,经计算淫羊藿素的得率为0.116%;淫羊藿(醇-酸体系)可以提取到淫羊藿素,其峰面积为4395.8,经计算淫羊藿素的得率为0.513%;淫羊藿(醇-酸体系+纤维素酶酶解)可以提取到淫羊藿素,其峰面积为
41791.8,经计算淫羊藿素的得率为5.116%。由图35~38可知,巫山淫羊藿(醇-水体系)无法提取到淫羊藿素;巫山淫羊藿(醇-水体系+纤维素酶酶解)可以提取到淫羊藿素,其峰面积为553.4,经计算淫羊藿素的得率为0.040%;巫山淫羊藿(醇-酸体系)可以提取到淫羊藿素,其峰面积为8197.7,经计算淫羊藿素的得率为0.981%;巫山淫羊藿(醇-酸体系+纤维素酶酶解)可以提取到淫羊藿素,其峰面积为11932.6,经计算淫羊藿素的得率为1.441%。
[0115]
根据两种淫羊藿药材提取处理液的液相结果可得:与醇-水体系提取相比,醇-酸水体系提取能使得淫羊藿素的得率提高,再经由纤维素酶酶解,其淫羊藿素的得率会明显高于前者。
[0116]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1