含有光亲和基团双吖丙啶的β-榄香烯衍生物及其制备方法和作为光亲和分子探针的应用
含有光亲和基团双吖丙啶的
β-榄香烯衍生物及其制备方法和作为光亲和分子探针的应用
技术领域
1.本发明属于光亲和分子探针的制备及应用领域,具体涉及一种含有光亲和基团双吖丙啶的β-榄香烯衍生物及其制备方法和作为光亲和分子探针的应用。
背景技术:
2.榄香烯(elemene)是从“浙八味”之一的温郁金块茎中提取出来的倍半萜类抗癌活性成分,临床上对肺癌、肝癌、脑胶质瘤等安全有效,并且对靶向药化疗药具有增敏减毒等优点。1994年国家药监局批准其为我国具有自主知识产权的抗肿瘤植物药。
3.新药研发的关键是深入理解药物在体内的作用机制并选择可以达到的最佳药效靶标。目前普遍认为榄香烯可通过干预多种信号转导途径发挥抗肿瘤作用。如抑制肿瘤细胞增殖,诱导癌细胞的凋亡,抑制肿瘤血管生成,抑制肿瘤细胞侵袭和转移,调节免疫功能等。然而很多研究尚停留在生物学表象变化的观察上,其具体分子机制及作用靶点的研究尚不明确。
4.化学分子探针是一类被赋予功能化的分子工具,能与感兴趣的研究对象通过共价键或非共价键结合,通过放射性、荧光、化学反应等手段监测,从而获得重要的生物大分子在细胞中的定位,作用靶标等信息。化学小分子探针的设计以充分的构效关系研究为基础,在不影响化合物活性的条件下,在适当的位置引入各个功能模块,实现不同检测的目的。一般的活性分子探针由两部分构成:含有反应基团的活性小分子,可以与靶标发生相互作用;报告基团,用于检测和分离小分子探针结合的生物靶标,包括荧光基团(用于生物靶标的可视化研究)、生物素(用于靶标分子的富集和纯化)或生物正交反应官能团(避免标记基团对活性小分子与生物靶标间相互作用的影响),如炔基或叠氮等。对于化学反应惰性且通过非共价作用力与靶标发生作用的活性小分子而言,为了减少非特异性吸附造成的假阳性,分子探针的设计中需额外引入一个光亲和基团,通过光照生成高活性的卡宾等中间体,与作用靶标发生共价交联,增加靶标筛选的范围。
5.很多著名的天然产物曾被成功地分子探针化并用于相应的分子作用机制、作用靶点和信号通路的研究中。例如,为了探究具有强烈细胞毒性的天然产物—肽类生物碱含氯环肽a的作用机理,harran课题组合成了生物素标记的含氯环肽,并应用它发现了其直接作用靶标是鸟氨酸氨基转移酶;天然产物ainsliatrimer是从菊科植物兔儿风的次级代谢产物愈创木内脂的三聚体,显示出对多种肿瘤细胞强烈的细胞毒性,然而其具体的作用机制仍不清楚。雷晓光课题组将天然产物全合成和生物正交相结合,利用天然产物分子探针化的策略,首次揭示了ppar为该天然产物的直接作用靶标。
6.榄香烯是只含有碳和氢两种元素的倍半萜类化合物,推测其与靶标分子的作用是非共价结合,结合力弱。因此发展榄香烯光亲和分子探针的构筑方法,对于探究榄香烯在生物体内的作用靶标的筛选、榄香烯构效关系的探究及榄香烯作用机制的研究很有必要。
技术实现要素:
7.本发明的第一个目的是针对现有技术的不足,提供一种含有光亲和基团双吖丙啶的β-榄香烯衍生物。
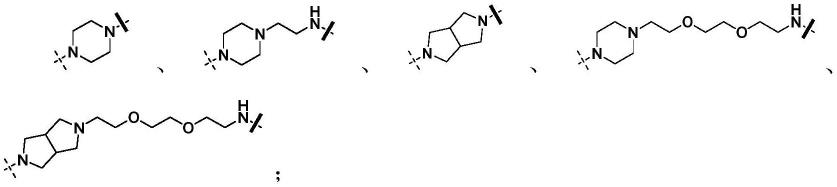
8.一种含有光亲和基团的β-榄香烯衍生物,或其光学异构体、消旋体、单一对映异构体、可能的非对映异构体,或其药学上可接受的盐、前药、氘代衍生物、水合物、溶剂化物,所述含有光亲和基团β-榄香烯衍生物的结构如式(i)所示:
[0009][0010]
其中:
[0011]
a为
[0012]
其中表示a和r连接的位点;
[0013]
r独立地选自以下结构片段之一:
[0014][0015]
其中表示r与榄香烯连接的位点,表示r与a连接的位点。
[0016]
作为优选,所述化合物的结构选自下组中的任意一种:
[0017][0018]
本发明的第二个目的是提供一种含有光亲和基团的β-榄香烯衍生物的制备方法,具体包括如下步骤:
[0019]
(1)将β-榄香烯(a-1)进行13位烯丙位溴取代反应得中间体a-2;
[0020]
(2)将含有氮杂原子官能团的r结构片段a-3通过选择性亲核取代反应连接到13位的β-榄香烯上,得中间体a-4;
[0021]
(3)对β-榄香烯13位上的结构片段去保护,得中间体a-5;
[0022]
(4)最后将a-5与探针片段与进行分子间酰胺缩合,得通式(i)所示的衍生物。
[0023]
其合成路线如下:
[0024][0025]
本发明式(i)所示化合物可通过如上的方法制得,然而该方法的条件,如反应物、溶剂、所用化合物的量、反应温度、反应所需时间等不限于上面的解释。本发明化合物还可以任选将在本说明书中描述的或本领域已知的各种合成方法组合起来而方便的制得,这样的组合可由本发明所属领域的技术人员容易地进行。
[0026]
本发明的第三个目的是提供上述含有光亲和基团双吖丙啶的β-榄香烯衍生物,或其光学异构体、消旋体、单一对映异构体、可能的非对映异构体,或其药学上可接受的盐、前药、氘代衍生物、水合物、溶剂化物在作为β-榄香烯光亲和分子探针上的应用。
[0027]
本发明的第四个目的是提供一种β-榄香烯相互作用蛋白筛选方法,采用上述含有光亲和基团双吖丙啶的β-榄香烯衍生物,或其光学异构体、消旋体、单一对映异构体、可能的非对映异构体,或其药学上可接受的盐、前药、氘代衍生物、水合物、溶剂化物作为光亲和分子探针。
[0028]
本发明具有以下优点:
[0029]
本发明通过在β-榄香烯骨架上引入光亲和基团(双吖丙啶)和报告基团(炔基)以及增加极性和水溶性基团后,制备得到具有光亲和活性的β-榄香烯衍生物,并合成了一系列的β-榄香烯光亲和分子探针。该β-榄香烯光亲和分子探针具有抑制各种肿瘤细胞株增殖的活性,经365nm波长照射后,能与细胞质内的蛋白质发生共价结合,能用于β-榄香烯相互作用蛋白的筛选,并有望用于榄香烯分子作用机制的探究。
附图说明
[0030]
图1为实施例10中β-榄香烯光亲和探针(化合物2)与对照化合物6和β-榄香烯的细胞成像结果。
具体实施方式
[0031]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0032]
实施例1:化合物1的制备
[0033][0034]
室温下,向化合物boc-l-谷氨酸-1-叔丁酯(100mg,0.33mmol)的dcm溶液中,在冰浴下依次加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(164.5mg,0.86mmol)、1-羟基苯并三唑(57.9mg,0.43mmol)和n,n-二异丙基乙胺(127.7mg,0.99mmol)。反应液在冰浴下搅拌1h,然后加入甲氧基甲基胺(41.8mg,0.43mmol)。反应液升至室温反应7h。减压除去dcm,剩余物再用乙酸乙酯(3
×
5ml)萃取。合并的有机相依次用水(2
×
5ml)和饱和食盐水(2
×
5ml)洗涤,并用无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(乙酸乙酯/石油醚体系)纯化,得到白色固体化合物1-1(103.5mg,产率90.7%)。1h nmr(500mhz,cdcl3)δ5.19(d,j=8.2hz,1h),4.16-4.21(m,1h),3.67(s,3h),3.17(s,3h),2.43-2.59(m,2h),2.10-2.19(m,1h),1.87-1.96(m,1h),1.45(s,9h),1.43(s,9h).
[0035]
在-78℃的条件下,向化合物1-1(360mg,1.04mmol)的thf(7.5ml)溶液中缓慢滴加1mol/l的memgbr溶液(4.68ml,4.68mmol)。反应液缓慢升至室温反应4h。加nh4cl溶液淬灭反应,减压除去thf,后处理同化合物1-1,得到白色固体化合物1-2(253.5mg,收率80.9%)。1h nmr(500mhz,cdcl3)δ5.06(s,1h),4.09(s,1h),2.41-2.57(m,2h),2.11(s,3h),2.05-2.06(m,1h),1.72-1.84(m,1h),1.42(s,9h),1.40(s,9h).
[0036]
在-78℃的条件下,化合物1-2(83.3mg,0.27mmol)溶于7mol/l的nh3/meoh(1.6ml,10.8mmol)溶液中搅拌7-8h。在-78℃的条件下加入羟胺磺酸(38.4mg,0.34mmol),反应液缓慢升至室温反应过夜,氨同时挥发。过滤除去白色固体并用甲醇(5ml
×
3)洗涤,母液浓缩至体积的一半。冰浴条件下,在母液中依次加入三乙胺和i2至反应液呈棕色,反应液升至室温搅拌1h,褪至无色。减压除去甲醇,后处理同化合物1-1,得到白色固体化合物1-3。1h nmr(500mhz,cdcl3)δ4.99(d,j=7.0hz,1h),4.13(d,j=5.3hz,1h),1.59(s,2h),1.43-1.44(m,18h,1.29-1.24(m,2h),1.00(s,3h).
[0037]
在冰浴条件下,化合物1-3(110.5mg,0.35mmol)溶于thf(4ml),缓慢加入4mol/l的hcl溶液。反应液在黑暗环境下搅拌过夜。减压除去thf和hcl溶液,析出白色固体化合物1-4。无需进一步纯化,直接用于下一步反应。1h nmr(500mhz,d
6-dmso)δ8.48(s,2h),3.79(s,1h),1.74-1.62(m,2h),1.48(ddt,j=85.0,15.3,8.2hz,2h),1.00(s,3h).
[0038][0039]
在冰浴条件下,化合物9-氨基-4,7-二氧杂壬酸叔丁酯(238mg,1.02mmol)溶于二氯甲烷(5ml)溶液中,加入4-戊炔酸(119.6mg,1.22mmol)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(216mg,1.13mmol)和4-二甲氨基吡啶(62.2mg,0.51mmol)。反应液升至室温反应7h。减压除去二氯甲烷,用乙酸乙酯(3
×
10ml)萃取,饱和食盐水洗涤(3
×
10ml),无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(二氯甲烷/甲醇体系)纯化,得到黄色油状化合物化合物1-5(343.4mg,85.5%)。1h nmr(500mhz,cdcl3)δ6.37(s,1h),3.72(t,j=6.3hz,2h),3.59(s,4h),3.56-3.53(m,2h),3.45(q,j=5.2hz,2h),2.54-2.47(m,4h),2.42(t,j=7.2hz,2h),1.98(t,j=2.6hz,1h),1.44(s,9h).
[0040]
在冰浴条件下,化合物1-5(216.7mg,0.69mmol)溶于二氯甲烷(5ml)中,缓慢加入甲酸(7ml)溶液。反应液升至室温搅拌8h。减压除去二氯甲烷和甲酸,得到化合物1-6(170mg,96%)。无需纯化,直接用于下一步反应。1h nmr(500mhz,cdcl3)δ6.54(s,1h),3.76(t,j=6.1hz,2h),3.61(q,j=5.2hz,4h),3.55(t,j=5.1hz,2h),3.45(q,j=5.3hz,2h),2.62(t,j=6.1hz,2h),2.54
–
2.49(m,2h),2.43(t,j=7.1hz,2h),2.00(t,j=2.6hz,1h).
[0041]
在冰浴条件下,化合物1-6(734mg,2.85mmol)溶于二氯甲烷(30ml)溶液中,加入n-羟基丁二酰亚胺(492.2mg,4.28mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(821.8mg,4.28mmol)和4-二甲氨基吡啶(174.5mg,1.43mmol)。反应液在室温下反应过夜。后处理同化合物1-5。得到黄色油状化合物1-7(863mg,82.3%)。1h nmr(500mhz,cdcl3)δ6.35(s,1h),3.85(t,j=6.1hz,2h),3.66
–
3.58(m,4h),3.57
–
3.52(m,2h),3.45(q,j=5.2hz,2h),2.91
–
2.80(m,6h),2.51(td,j=7.4,6.8,2.5hz,2h),2.40(t,j=7.2hz,2h),1.99(t,j=2.6hz,1h).
[0042]
在室温条件下,在化合物1-4(196mg,1.25mmol)的甲醇(4ml)溶液中加入化合物1-7(441.9mg,1.25mmol)和三乙胺(378.8mg,3.75mmol),在黑暗环境下反应2h。减压除去三乙胺和甲醇,剩余物加水溶解,用1mol/l hcl溶液调ph值至5。用乙酸乙酯(3
×
5ml)萃取水溶液3次,收集有机相,饱和食盐水洗涤,无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(二氯甲烷/甲醇体系)纯化,得到淡黄色油状化合物1-8(288mg,收率58.3%)。1h nmr(500mhz,cdcl3)δ7.03(d,j=7.7hz,1h),6.75(s,1h),4.55(td,j=7.5,5.6hz,1h),3.73(d,j=15.3hz,2h),3.61(s,4h),3.55(d,j=6.2hz,2h),3.49
–
3.40(m,2h),2.53
–
2.50(m,4h),2.45(t,j=6.9hz,2h),2.02(t,j=2.6hz,1h),1.86
–
1.78(m,1h),1.62
–
1.55(m,1h),1.51
–
1.44(m,1h),1.37(ddd,j=14.6,11.5,5.1hz,1h),
1.01(s,3h).
[0043][0044]
室温条件下,向榄香烯13位溴代化合物(162mg,0.68mmol)的超干dmf(4ml)溶液中,加入1-boc-哌嗪(153mg,0.82mmol)及真空干燥的碳酸铯(332mg,1.02mmol)。反应液逐渐升至60℃搅拌4h。减压除去dmf,剩余混合物用二氯甲烷(3
×
5ml)萃取。合并的有机相依次用水(3
×
5ml)和饱和食盐水(3
×
5ml)洗涤,并用无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(纯石油醚洗脱)纯化,得到无色油状化合物1-9(189mg,收率71.7%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.3,11.0hz,1h),4.91(m,4h),4.83-4.80(m,1h),4.58(s,1h),3.46-3.35(m,4h),2.91(q,j=13.4hz,2h),2.32(s,4h),2.08(m,1h),2.02(dd,j=12.1,4.0hz,1h),1.71(s,3h),1.64-1.47(m,6h),1.45(s,9h),1.01(s,3h).
[0045]
将1-9(189mg,0.488mmol)溶于干燥二氯甲烷(1.5ml)。于0℃下加入三氟乙酸(1.5ml),逐渐升至室温搅拌3h。缓慢滴加碳酸钾(4ml)及水(5ml)淬灭反应。减压除去三氟乙酸和溶剂,剩余物用乙酸乙酯稀释(20ml),用饱和碳酸钾溶液(3ml)洗涤,水层反萃一次(10ml),合并的有机相用饱和食盐水洗(3ml),并用无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩后抽干得黄色油状化合物1-10(120mg,收率85.2%)。
[0046]
在冰浴条件下,化合物1-8(46.3mg,0.12mmol)溶于dmf(2ml)溶液中,加入1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(59.4mg,0.31mmol)、1-羟基苯并三唑(21.6mg,0.16mmol)和n,n-二异丙基乙胺(46.4mg,0.36mmol),反应液搅拌1h后。加入化合物1-10(45.8mg,0.12mmol)。反应液在黑暗的室温条件下反应6h。后处理同化合物1-5,得到淡黄色油状化合物1(26.1mg,32.6%)。1h nmr(500mhz,cdcl3)δ7.13(d,j=8.3hz,1h),7.04(s,1h),5.81(dd,j=17.3,11.0hz,1h),4.98
–
4.87(m,5h),4.82(s,1h),4.58(s,1h),3.75
–
3.59(m,8h),3.59
–
3.49(m,6h),2.93(q,j=13.4hz,2h),2.54
–
2.48(m,4h),2.45
–
2.33(m,6h),2.06
–
1.99(m,2h),1.97(t,j=2.6hz,1h),1.70(s,3h),1.61
–
1.34(m,10h),1.00(d,j=2.6hz,6h).
[0047]
实施例2:化合物2的制备
[0048][0049]
参照实施例1的合成方法,第一步反应得到黄色油状化合物2-1(537mg,76.3%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.4,10.9hz,1h),4.94-4.87(m,4h),4.83-4.80(m,1h),4.58(s,1h),3.22(d,j=4.6hz,2h),2.91(q,j=13.4hz,2h),2.55-2.29(m,10h),2.10-2.05(m,1h),2.02(dd,j=12.3,3.8hz,1h),1.71(s,3h),1.65-1.47(m,6h),1.44(s,9h),1.01(s,3h).
[0050]
第二步反应黄色油状化合物2-2(321.7mg,78.2%)。第三步反应黄色油状化合物2(119.2mg,48.5%)。1h nmr(500mhz,cdcl3)δ7.10(d,j=8.4hz,2h),6.71(s,1h),5.81(dd,,j=17.4,10.9hz,1h),4.90(dd,j=17.2,10.2hz,4h),4.83
–
4.78(m,1h),4.57(s,1h),4.41(q,j=7.2,6.4hz,1h),3.74
–
3.69(m,2h),3.60(s,4h),3.58
–
3.31(m,,6h),2.97
–
2.87(m,2h),2.54
–
2.36(m,16h),2.06
–
2.01(m,2h),1.99
–
1.97(m,1h),1.70(s,3h),1.64
–
1.52(m,4h),1.46(ddd,j=23.4,13.0,6.0hz,4h),1.31
–
1.27(m,2h),1.01(d,j=7.6hz,6h).
[0051]
实施例3:化合物3的制备
[0052][0053]
室温下,向化合物2-[2-(2-t-boc-氨基乙氧基)乙氧基]乙醇(44mg,0.177mmol)的dcm(2ml)溶液中,依次加入三乙胺(35.7mg,0.354mmol)、对甲苯磺酰氯(40.5mg,0.212mmol)、4-二甲氨基吡啶(2.12mg,0.018mmol)。反应液室温搅拌7h。用饱和nacl(3
×
5ml)洗涤,无水na2so4干燥,过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(二氯甲烷/甲醇体系)纯化,得到无色油状化合物3-1(67.2mg,收率94.4%)。1h nmr(400mhz,cdcl3)δ7.79(d,j=8.3hz,3h),7.34(d,j=8.1hz,3h),4.94(s,1h),4.16-4.17(m,2h),3.68-3.70(m,2h),3.56-3.58(m,2h),3.53-3.55(m,2h),3.48-3.50(m,2h),3.28-3.29(m,2h),2.44(s,1h),1.43(s,9h)。
[0054]
室温下,向化合物3-1(166.8mg,0.414mmol)的ch3cn(2ml)溶液中加入苄基-1-哌嗪碳酸酯(139.2mg,0.633mmol)、真空干燥的碳酸铯(168mg,0.515mmol)和ki(22mg,0.15mmol)。反应液逐渐升至80℃搅拌7h。减压除去ch3cn,剩余混合物用二氯甲烷(3
×
5ml)萃取。合并的有机相依次用水(3
×
5ml)和饱和食盐水(3
×
5ml)洗涤,并用无水硫酸钠干燥。过滤除去干燥剂,滤液在减压条件下浓缩,所得的粗品经硅胶柱层析(二氯甲烷/甲醇体系)纯化,得到无色油状化合物3-2(508mg,收率45.1%)。1h nmr(400mhz,cdcl3)δ7.34(d,j=3.9hz,5h),5.29(s,1h),5.12(s,2h),3.60(d,j=5.6hz,6h),3.54
–
3.49(m,6h),3.30(q,,j=4.9hz,2h),2.60(t,j=5.7hz,2h),2.46(s,4h),1.43(s,9h).
[0055]
室温下,向化合物3-2(59.3mg,0.132mmol)的meoh(2ml)溶液中加入10%pd/c(19.2mg),在h2条件下反应过夜,过滤除去10%pd/c,滤液在减压条件下浓缩,得化合物3-3(35mg,84%),无需柱层析,直接下一步反应。
[0056]
参照实施例1中化合物1-9的合成方法,反应得到无色油状化合物3-4(191.7mg,收率70.2%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.4,10.8hz,1h),4.93
–
4.87(m,4h),4.82
–
4.79(m,1h),4.58(s,1h),3.60(d,j=6.9hz,6h),3.53(t,j=5.0hz,2h),3.30(t,j=5.4hz,2h),2.95
–
2.86(m,2h),2.62
–
2.40(m,8h),2.01(dd,j=12.1,3.9hz,2h),1.70(s,3h),1.66
–
1.45(m,6h),1.44(s,9h),1.00(s,3h).
[0057]
参照实施例1中化合物1-10的合成方法,反应得黄色油状化合物3-5(132.9mg,收率85.9%)。
[0058]
参照实施例1中化合物1的合成方法,反应得到黄色油状化合物3(89.3mg,35.6%)。1h nmr(500mhz,cdcl3)δ7.66(s,1h),7.36(d,j=7.3hz,1h),7.14(s,1h),5.82(dd,j=17.8,10.5hz,1h),5.05
–
4.95(m,2h),4.92(d,j=4.5hz,1h),4.89(s,1h),4.84
–
4.81(m,1h),4.58(s,1h),4.17
–
4.10(m,1h),3.76
–
3.68(m,3h),3.63
–
3.59(m,8h),3.58
–
3.55(m,4h),3.52(dq,j=6.6,3.2,2.7hz,2h),3.41(dd,j=11.0,7.8hz,3h),3.05(s,2h),2.84(s,6h),2.53(td,j=7.3,6.7,3.7hz,4h),2.46(t,j=7.0hz,2h),2.13(d,j=22.8hz,4h),2.07(s,1h),2.05(s,1h),1.99(d,j=2.5hz,1h),1.71(s,3h),1.59
–
1.39(m,10h),1.01(d,j=8.0hz,6h).
[0059]
实施例4:化合物4的制备
[0060][0061]
参照实施例1中化合物1-9的合成方法,反应得到黄色油状化合物4-1(113.87mg,67.6%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.3,11.0hz,1h),4.93(s,1h),4.90(dd,j=7.6,1.3hz,1h),4.88(s,1h),4.85(s,1h),4.82-4.80(m,1h),4.58(s,1h),3.55(s,2h),3.19(s,2h),2.99(q,j=15hz,2h),2.76(s,2h),2.54(s,2h),2.39(d,j=7.1hz,2h),2.10-1.99(m,2h),1.70(s,3h),1.67-1.48(m,6h),1.45(s,9h),1.00(s,3h).
[0062]
参照实施例1中化合物1-10的合成方法,反应得到黄色油状化合物4-2(77.6mg,90%)。
[0063]
参照实施例1中化合物1的合成方法,反应得到黄色油状化合物4(38.3mg,32.6%)。lcms:m/z 693.5[m+h]
+
。1h nmr(500mhz,cdcl3)δ7.17(ddd,j=45.8,12.9,6.9hz,2h),5.82(ddd,j=17.4,10.9,3.5hz,1h),4.97
–
4.86(m,4h),4.82(dt,j=5.0,1.8hz,1h),4.74(td,j=7.9,4.6hz,1h),4.58(dd,j=4.6,2.0hz,1h),3.71(q,j=5.9hz,4h),3.65
–
3.51(m,10h),3.08
–
2.95(m,2h),2.63
–
2.45(m,12h),2.05
–
1.99(m,3h),1.98(t,j=2.6hz,1h),1.71(d,j=4.2hz,3h),1.61
–
1.35(m,12h),1.02
–
0.98(m,6h).
[0064]
实施例5:化合物5的制备
[0065][0066]
参照实施例3中化合物3-2的制备,反应得到化合物5-1(56.8mg,收率45.1%)。1h nmr(500mhz,cdcl3)δ7.36-7.27(m,5h),5.11(s,3h),3.59(s,6h),3.57(d,j=5.9hz,2h),3.52(t,j=5.1hz,2h),3.40-3.26(m,4h),2.82(s,4h),2.65(t,j=5.9hz,2h),2.36(s,2h),1.43(s,9h)。
[0067]
参照实施例3中化合物3-3的制备,反应得化合物5-2,无需柱层析,直接用于下一步反应。
[0068]
参照实施例1中化合物1-9的合成方法,反应得到黄色油状化合物5-3(240mg,72.8%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.4,10.9hz,1h),4.94-4.87(m,4h),4.83-4.80(m,1h),4.58(s,1h),3.22(d,j=4.6hz,2h),2.91(q,j=13.4hz,2h),2.55-2.29(m,10h),2.10-2.05(m,1h),2.02(dd,j=12.3,3.8hz,1h),1.71(s,3h),1.65-1.47(m,6h),1.44(s,9h),1.01(s,3h).
[0069]
参照实施例1中化合物1-10的合成方法,反应得到黄色油状化合物5-4(188.1mg,96%)。
[0070]
参照实施例1中化合物1的合成方法,反应得到黄色油状化合物5(57.5mg,30.2%)。1h nmr(500mhz,cdcl3)δ7.25(d,j=7.6hz,2h),7.08(s,1h),5.81(dd,j=17.4,10.9hz,1h),4.96
–
4.85(m,4h),4.83
–
4.78(m,1h),4.58(s,1h),4.54
–
4.42(m,1h),3.72(dq,,j=8.7,4.7,4.2hz,4h),3.64
–
3.51(m,13h),3.47
–
3.36(m,3h),3.07
–
3.00(m,2h),2.84(s,5h),2.62
–
2.39(m,9h),2.38
–
2.19(m,4h),2.00
–
1.98(m,1h),1.70(s,3h),1.63
–
1.42(m,8h),1.28(d,j=5.5hz,4h),1.00(s,6h).
[0071]
实施例6:对照化合物6的制备
[0072][0073]
参照实施例1中化合物1-9的合成方法,反应得到黄色油状化合物6(113.6mg,62.8%)。1h nmr(500mhz,cdcl3)δ5.82(dd,j=17.5,10.9hz,1h),4.94
–
4.87(m,4h),4.81(p,j=1.6hz,1h),4.58(d,j=2.0hz,1h),2.96
–
2.86(m,2h),2.41(s,8h),2.28(s,3h),2.01(dd,j=12.3,3.9hz,2h),1.70(d,j=2.3hz,3h),1.64
–
1.40(m,6h),1.00(s,3h).
[0074]
实施例7:对照化合物7的制备
[0075][0076]
室温条件下,向顺式-2-boc-六氢吡咯并[3,4-c]吡咯化合物(212.3mg,2.35mmol)的dcm(10ml)溶液中,加入乙酰氧基硼氢化钠(1.5g,7.1mmol)及甲醇溶液(355mg,11.8mmol)。室温反应过夜,加入k2co3淬灭反应,调ph至9左右,后处理同化合物1-1。得到无色油状化合物7-1(523mg,收率97.6%)。1h nmr(500mhz,cdcl3)δ3.53(s,2h),3.22(s,2h),2.80(dh,j=9.3,4.0hz,2h),2.65
–
2.59(m,2h),2.36(d,j=9.0hz,2h),2.31(s,3h),1.44(s,9h).
[0077]
参照实施例1中化合物1-10的合成,反应得到化合物7-2(103mg,92%)。
[0078]
参照实施例1中化合物1-9的合成,反应得到化合物7(56.3mg,53.6%)。1h nmr(500mhz,cdcl3)δ5.85
–
5.76(m,1h),5.05
–
4.77(m,5h),4.58(d,j=2.1hz,1h),3.17
–
3.04(m,3h),2.97
–
2.87(m,2h),2.59
–
2.38(m,8h),2.09
–
1.96(m,2h),1.71(d,j=2.0hz,3h),1.66
–
1.17(m,8h),1.00(s,3h).
[0079]
实施例8:对照化合物8的制备
[0080][0081]
参照实施例1中化合物1的合成方法,反应得到黄色油状化合物8(57.5mg,
40.6%)。1h nmr(400mhz,chloroform-d)δ4.90(td,j=7.8,4.4hz,1h),3.74
–
3.56(m,14h),2.46
–
2.37(m,8h),2.33(s,5h),1.98(t,j=2.6hz,1h),1.67(s,2h),1.25(s,2h),1.01(s,3h).
[0082]
实施例9:体外抗肿瘤活性评价(肿瘤细胞增殖抑制实验)
[0083]
1实验设备与试剂
[0084]
1.1仪器
[0085]
生物安全柜(上海百基生物科技有限公司)、恒温二氧化碳培养箱(thermo)、酶联免疫分析仪(spark)、倒置显微镜(nikon)、移液枪一套(eppendorf)和离心机(beckman coulter)。
[0086]
1.2试剂
[0087]
dmem(bi)、rpmi 1640(bi)、fatal bovine serum(excell)、pbs(浙江森瑞生物科技有限公司)、trypsin(浙江森瑞生物科技有限公司)、dmso(coolaber)和cck-8(coolaber)、双抗(gbico)。
[0088]
1.3细胞株
[0089]
人肝癌细胞huh1、人肺癌细胞a549、人脑胶质瘤细胞u87-mg。
[0090]
2实验方法
[0091]
2.1细胞复苏及常规培养
[0092]
(1)细胞实验室进行常规消毒,超净工作台紫外照射30min以上。
[0093]
(2)培养液(dmem)、0.25%胰蛋白酶(trypsin)恒温水浴箱37℃预热20min,二甲基亚砜(dmso)在4℃冰箱中冷藏30min。
[0094]
(3)从液氮保存罐中取出冻存管,立即放入37℃水浴中,快速摇晃,直至冻存液完全融化。
[0095]
(4)将细胞悬液移入离心管中,缓慢加入培养液,移至离心机中离心(120
×
g,5min)。
[0096]
(5)加入培养液调整细胞浓度,摇匀后放置于37℃、5%co2培养箱中培养。
[0097]
(6)细胞贴壁24h后换培养液,待细胞密度达到80%-90%用胰酶消化后传代。
[0098]
2.2肿瘤细胞增殖抑制实验方法
[0099]
(1)用dmem培养基将a549细胞悬液调整到5
×
104/ml。每孔加100μl细胞悬液于96孔细胞培养板,最终细胞浓度为5000细胞/孔,放置于37℃、5%co2培养箱中培养24h。
[0100]
(2)用完全培养基将榄香烯原料药稀释至所需浓度,移除孔内培养基,每孔分别加入100μl含药的培养基,每个药物浓度各3个复孔,同时设置对照组以及空白组。
[0101]
(3)将96孔板置于37℃,5%co2孵箱中培养48h。
[0102]
(4)药物处理48h后,将培养液吸出,每孔加入含有10%cck-8的100μl培养液,培养箱内继续培养1h后用thermo fisher multiskan fc酶标仪测定450nm吸光度。
[0103]
(5)应用ibm spss statistics 22.0软件,使用非线性回归模型绘制s型剂量-存活率曲线并用以下公式计算存活率和抑制率:
[0104]
细胞存活率=[(as-ab)/(ac-ab)]
×
100%
[0105]
抑制率=[(ac-as)/(ac-ab)]
×
100%
[0106]
as:实验孔(含有细胞的培养基、cck-8、待测药物)的吸光度
[0107]
ac:对照孔(含有细胞的培养基、cck-8、没有待测药物)的吸光度
[0108]
ab:空白孔(不含细胞和待测药物的培养基、cck-8)的吸光度
[0109]
3实验结果
[0110]
按上述实验方法使用cck-8测定了分离纯化得到的β-榄香烯单体和榄香烯光亲和探针及对照化合物对三种人癌细胞的增殖抑制作用,即a549人非小细胞肺癌细胞、u87-mg人恶性脑胶质瘤细胞、huh1人肝癌细胞,结果见表1。
[0111]
表1β-榄香烯、β-榄香烯光亲和探针(化合物1-5)及对照化合物(化合物6-8)的ic
50
[0112][0113]
注明:nd表示未检测到抗肿瘤活性
[0114]
表1中的体外抗肿瘤活性结果表明,测定的7个化合物对人a549肺癌细胞、人huh1肝癌细胞和人u87-mg脑胶质瘤细胞的抑制活性均强于β-榄香烯。对于人a549肺癌细胞,有3个化合物(1、2、5)的ic
50
值比β-榄香烯低5倍以上;对人huh1肝癌细胞,有5个化合物(1、2、3、5、7)的ic
50
值比β-榄香烯低10倍以上;对人u87-mg恶性脑胶质瘤细胞,有2个化合物(3、5)的ic
50
值比β-榄香烯低10倍以上。以上结果表明,在保证β-榄香烯骨架完整的基础上,通过引入含氧原子、氮原子的极性基团和具有增强水溶性的peg链增强了β-榄香烯的体外抗肿瘤活性。
[0115]
实施例10:在活细胞中使用分子探针进行预靶向成像研究
[0116]
为了判断分子探针是否可以进入胞内,并探究其靶向的生物大分子的结合部位,按如下方法进行实验。
[0117]
1实验设备与试剂
[0118]
1.1仪器
[0119]
便携式led紫外线探伤灯(luyor)、激光共聚焦显微镜(olympus,fv3000rs)、恒温二氧化碳培养箱(thermo)
[0120]
1.2试剂
[0121]
dmem培养基、pbs、4%多聚甲醛组织细胞固定液、triton x-100、5%bsa封闭液、dapi染色液、cu2so4、抗坏血酸钠、thpta、n
3-tamra
[0122]
2实验方法
[0123]
(1)105的huh1肝癌细胞接种于玻底培养皿中,于37℃、5%co2培养箱中培养至细胞贴壁;
[0124]
(2)加入含不同药物浓度的dmem培养基,37℃、5%co2培养箱中孵育过夜;
[0125]
(3)移除含药的培养基,用预冷的pbs(3
×
1ml)洗涤3min,置于冰上,365nm波长下照射15min;
[0126]
(4)4%多聚甲醛组织细胞固定液1ml处理15min,移除后用预冷的pbs洗涤;
[0127]
(5)用500μl的0.1%triton x-100的pbs溶液透膜10min,再用1ml的5%bsa封闭液封闭30min,移除后用预冷的pbs洗涤(3
×
1ml);
[0128]
(6)加入200μl新鲜配置的click反应液(20μm n
3-tamra、400μm cu2so4、400μm thpta、2mm抗坏血酸钠),在室温下震荡孵育2h移除反应液,用预冷的pbs(3
×
1ml)洗涤3min;
[0129]
(7)加入200μldapi染色液,染色6min,移除后用预冷的pbs(3
×
1ml)洗涤3min,用激光扫描共聚焦显微镜成像。
[0130]
4实验结果
[0131]
按上述实验方法测定了β-榄香烯光亲和探针(化合物2)与对照化合物6和β-榄香烯的细胞成像,结果如图1所示。
[0132]
结果表明,加入dmso或加入化合物2(20μm)且不进行uv照射时,胞内无罗丹明红色荧光;加入化合物2(20μm)并进行uv照射时,胞内有强烈的罗丹明红色荧光;加入化合物2(20μm)和化合物6(20μm)、化合物2(20μm)和β-榄香烯(50μm)进行uv照射时,胞内罗丹明红色荧光强度大幅减弱。以上结果表明,β-榄香烯光亲和探针(化合物2)与靶标的结合为非共价结合;化合物6和β-榄香烯均能与化合物2发生竞争性结合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1