一种制备瑞马唑仑关键中间体的方法与流程
1.本发明属于医药技术领域,具体涉及一种阵痛麻醉药瑞马唑仑的关键中间体的制备方法。
背景技术:
2.瑞马唑仑(remimazolam)是一种超短效静脉注射苯二氮卓镇静剂/麻醉剂,作用于gaba-α受体。在人体中,remimazolam被组织酯酶迅速代谢成无活性代谢物,并且不被细胞色素依赖性肝脏途径代谢,是一种超短效苯二氮卓类药物,其作为静脉全身麻醉药物,具有起效快、持续时间短、苏醒快和耐受性良好的特点。瑞马唑仑用于麻醉诱导、麻醉维持和日间手术麻醉,相比其他产品在应用于伴有心血管疾病、呼吸系统疾病、肝病以及老年患者时具有一定的优势。
3.苯磺酸瑞马唑仑于2020年7月正式获批上市,是用于无痛诊疗镇静、全身麻醉、icu镇静以及局麻镇静等领域。苯磺酸瑞马唑仑是一种新型的超短效镇静麻醉药物,与其他同类产品相比,苯磺酸瑞马唑仑起效更快,代谢迅速且代谢产物活性低,可以减少药物之间的相互作用。该药物的出现可能会重塑麻醉用药的格局。
4.wo0069836a1和wo2013029431a1公开了一种苯并二氮杂衍生物及其托西酸盐的制备方法,具体方法如下:该方法在制备化合物3时所用反应物需要在偶合剂作用下下发生偶合反应,在碱性条件下闭环反应,并且需要加入酸脱保护基fmoc,总收率仅为48.2%,收率偏低。反应方程式如下:
[0005][0006]
wo2011032692a1公开了另一种苯并二氮杂衍生物的制备方法,具体方法如下:
[0007][0008]
该方法在制备化合物4时起始反应物为n-boc-glu(ome)-oh,在偶合剂dcc作用下反应得到化合物2,再加入盐酸脱boc保护基得到化合物3,加入碳酸氢钠环化反应得到化合物4。三步总收率67%,产物化学纯度98.35%。化合物4是粘性油状物,用异丙醇加热溶解,冷却析晶,过滤得到黄色晶体。得到化合物2和3都是粗品直接投下一步,有很多杂质累积到了化合物4这步,导致化合物4纯度不高,影响产品质量。用该工艺得到的中间体得到瑞马唑仑化学纯度为93.91%,产物纯度较低。
[0009]
wo2019/72944公开了保护基为cbz时制备方法,文献中产品纯度为99%,但无手性纯度数据,本发明实施例1重复了该实验,仅有96.5%ee。然而由于该产品手性纯度后续提高手段有限,例如采用重结晶等进行提高时,收率损失较大,而且需要多次重结晶才能达到99.8%以上。
[0010]
瑞马唑仑作为医药产品,对药品纯度要求很高,关键中间体纯度也很重要。因此,有必要优化工艺,开发出制备高纯度瑞马唑仑中间体的方法。
技术实现要素:
[0011]
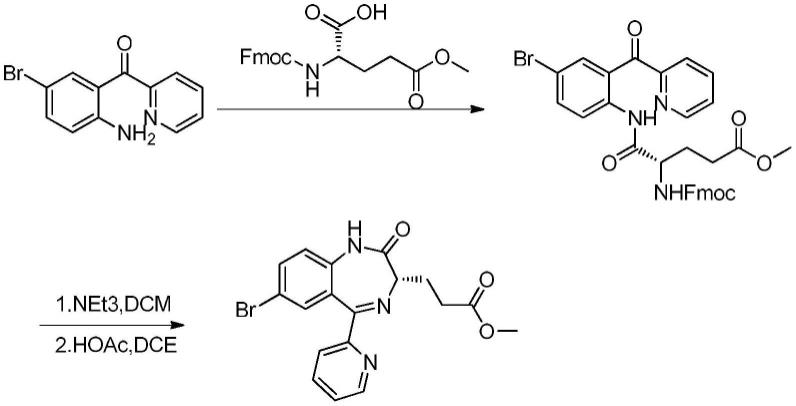
本发明提供了一种瑞马唑仑关键中间体d的改进制备方法,采用2-(2-氨基-5-溴-苯甲酰基)吡啶a和n-tr-谷氨酸-5-甲酯为原料,在含硼试剂作用下缩合反应得到化合物b,再脱保护得到化合物c,最后碱性低温条件下关环得到得到化合物d。该方法工艺重现性好,操作简便稳定、各步产物容易分离、收率高、环境友好、适合工业化规模生产的。
[0012]
本发明提供的一种瑞马唑仑关键中间体的制备方法,包括以下步骤:从2-(2-氨基-5-溴-苯甲酰基)吡啶a出发,与n-tr-谷氨酸-5-甲酯缩合得到中间体b;接着中间体b脱保护,得到中间体c;中间体c经过关环反应得到化合物d。合成路线如下:
[0013][0014]
本发明所述技术方法,采用具体步骤如下:
[0015]
第一步:中间体b的合成
[0016]
从2-(2-氨基-5-溴-苯甲酰基)吡啶a出发,与n-tr-谷氨酸-5-甲酯e在催化剂存在下,有机溶剂中缩合反应得到中间体b;
[0017]
进一步地,所述缩合试剂选自硼酸三(三氟乙醇)酯、3,5-二硝基苯硼酸或(c6f5)3b。
[0018]
进一步地,所述有机溶剂选自四氢呋喃、二氧六环、甲苯、环己烷、正己烷、环丁砜中的一种或多种。
[0019]
进一步地,所述2-(2-氨基-5-溴-苯甲酰基)吡啶a与n-tr-谷氨酸-5-甲酯e摩尔比为1:1-1.2。
[0020]
第二步:化合物d的合成
[0021]
中间体b在有机溶剂中,酸性条件下脱tr保护,得到中间体c,然后在碱性低温条件下关环得到产物d。
[0022]
进一步地,有机溶剂选自四氢呋喃、二氧六环、dmso、dmf、2-甲基四氢呋喃、环丁砜中的一种或多种。
[0023]
进一步地,所述酸选自三氟醋酸、盐酸、氯化氢等。
[0024]
进一步地,所述碱选自有机碱,例如dbu、吗啉、n-甲基吗啉、吡啶、三乙胺等。其中采用n-甲基吗啉和吗啉时,给出最好反应对应选择性,反应时间通常可在2小时内完成。
[0025]
进一步地,所述低温条件为-10℃至0℃下进行。
[0026]
采用常见无机碱,例如碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾等时,例如参考wo2011032692a1重复实验时,随着反应进行,反应对映选择性降低,最终对映选择性达到97-99%之间,调整反应温度也不能达到99.5%以上。采用降低反应温度至0℃以下,反应速度明显变慢,进行48小时后,仍有不少于8%以上原料剩余,增加碱当量时,对应选择性明显下降。
[0027]
本发明有益效果
[0028]
第一、缩合反应时,采用tr保护谷氨酸甲酯和硼试剂脱水,相比传统dcc缩合,副产物容易除去,且回流脱水过程中对映选择性无变化;
[0029]
第二、脱保护和关环时,脱保护酸性条件下进行,关环在有机碱/低温条件下进行(采用吗啉或n-甲基吗啉在0℃至-10℃反应),游离脱保护后中间体的同时进行关环,手性
中心无消旋化现象。
附图说明
[0030]
图1为实施例4化合物d的第一次中控的手性hplc谱图;
[0031]
图2为实施例4化合物d的第二次中控的手性hplc谱图;
[0032]
图3为实施例4化合物d纯化后的手性hplc谱图;
具体实施方式
[0033]
下面结合具体实施例,进一步阐述本发明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。
[0034]
实施例1
[0035]
第一步:
[0036]
将2-(2-氨基-5-溴-苯甲酰基)吡啶(38.6g,139mmol)和n-cbz谷氨酸-5-甲酯(45.2g,153mmol)在15℃下加入二氯甲烷(200ml)中,然后溶液冷却至-10℃。将n,n'-二环己基碳二亚胺(32.2g,156mmol)的二氯甲烷(65ml)溶液在-10℃下缓慢加入上述溶液中,该反应在-10℃搅拌48小时,反应液升温至15℃过滤。滤液低于25℃减压蒸馏,然后加入250ml甲基叔丁基醚,溶液加热至50℃后慢慢冷却至25℃,过滤50℃烘干得浅黄色固体(72.3g,收率:93.6%)。
[0037]
第二步:
[0038]
将中间体(35g,63mmol)加入冰醋酸(70ml)中,将33%溴化氢/冰醋酸溶液(45.7ml,253mmol)在10-12℃下慢慢加入上述反应液中,加完升温至20℃,并在15-20℃下搅拌2小时。加入水(120ml)和二氯甲烷(50ml),分离水相,控制温度在25℃采用碳酸氢钠调节ph=3.8-4,用二氯甲烷萃取,有机相减压蒸馏,加入50ml异丙醇并加热至82℃,加入50ml正庚烷,慢慢冷却至20℃,过滤烘干得22.4g,收率88.2%,hplc 99.83%,96.5%ee。
[0039]
实施例2
[0040]
第一步:
[0041]
将n-tr-谷氨酸-5-甲酯(1.46g,3.61mmol)、2-(2-氨基-5-溴-苯甲酰基)吡啶a(1.0g,3.61mmol)和二氧六环8ml/环己烷25ml混合,搅拌均匀。接着加入b(och2cf3)3(0.22g,0.72mmol)升温回流分水反应8小时,tlc检测反应完毕。减压浓缩,加入乙酸乙酯和水萃取反应。饱和食盐水洗涤,有机层旋蒸得到粗品,经甲醇/水重结晶得中间体b2.22g,收率93.0%。
[0042]
第二步:
[0043]
将氯化氢气体通入b(1.46g,2.2mmol)的四氢呋喃(15ml)溶液中15分钟,tlc显示脱完保护后,降温至-5℃,然后开始滴加吗啉(0.96g,11mmol),滴加完后保持低温反应2小时。用二氯甲烷萃取,有机相减压蒸馏,加入10ml甲基叔丁基醚,慢慢冷却至20℃,过滤烘干得0.81g,收率91.4%,hplc:99.43%,ee:99.92%。
[0044]
实施例3
[0045]
第一步:
[0046]
将n-tr-谷氨酸-5-甲酯e(1.2kg,2.99mol)和2-(2-氨基-5-溴-苯甲酰基)吡啶a(0.69kg,2.49mol)加入四氢呋喃5l/甲苯11l溶剂中,搅拌均匀。接着加入b(c6f5)3(61.4g,0.12mol),升温至回流分水反应6小时,tlc检测反应完毕。减压浓缩,加入乙酸乙酯和水萃取反应。有机层用食盐水洗涤,干燥旋干得到粗品,经甲醇/水重结晶得中间体b1.5kg,收率91.5%。
[0047]
第二步:
[0048]
将中间体b(1.5kg,2.26mol)和醋酸(0.2kg,3.39mol)加入二氧六环(8l)中,tlc显示脱完保护后,降温至-10℃,然后开始滴加三乙胺(1.14kg,11.3mmol),滴加完后保持低温反应2小时。用二氯甲烷萃取,有机相减压蒸馏,加入10l甲基叔丁基醚,慢慢冷却至20℃,过滤烘干得0.84kg,收率92.3%,hplc 99.84%,99.42%ee。
[0049]
实施例4
[0050]
第一步:
[0051]
将n-ns-谷氨酸-5-甲酯e(0.1kg,0.29mol)和2-(2-氨基-5-溴-苯甲酰基)吡啶a(0.088kg,0.32mol)加入四氢呋喃0.3l/甲苯0.6l溶剂中,搅拌均匀。接着加入b(c6f5)3(6.14g,0.012mol),升温至回流分水反应6小时,tlc检测反应完毕。减压浓缩,加入乙酸乙酯和水萃取反应。有机层用食盐水洗涤,干燥旋干得到粗品,经甲醇/水重结晶得中间体0.15kg,收率85.71%。
[0052]
第二步:
[0053]
将中间体(0.15kg,0.25mol)、三乙胺(76.28g,0.75mol)和苯硫醇(33g,0.3mol)加入n,n-二甲基甲酰胺(1.5l)中,60℃搅拌1小时,取样送检98.92%ee(图1)。继续搅拌1小时反应完成。加入水,用二氯甲烷萃取,有机相减压蒸馏,加入1l甲基叔丁基醚,慢慢冷却至20℃,过滤烘干得91.84g,收率91.60%,hplc:99.82%,98.08%ee(图2)。取上述粗品(50g)加入于50ml异丙醇中,打浆搅拌4小时,过滤,干燥得目标化合物16.21g,收率32.42%,99.08%ee(图3)。
[0054]
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1