一类香豆素衍生物及其制备方法
1.本发明涉及香豆素衍生物技术领域,具体为一类香豆素衍生物及其制备方法。
背景技术:
2.目前以简单原料温和高效地合成烯烃类香豆素类化合物报道仍然较少。例如要求苛刻的反应条件强酸等、毒性试剂(tbab等)、以及高活性碘代物的使用。并且现有路线步骤繁琐,起始原料不通用,反应时间长,原子经济性不好,制备成本高,总产率低;并且heck偶联过程中用到过量的tbab,不仅后处理困难,而且对于环境也有一定的污染;鉴于此,发展简易经济的合成方法以合成多样的烯烃类香豆素类化合物是非常重要的。
3.基于此,本发明设计了一类香豆素衍生物及其制备方法,以解决上述问题。
技术实现要素:
4.本发明的目的在于提供一类香豆素衍生物及其制备方法,以解决上述背景技术中提出的问题。
5.为实现上述目的,本发明提供如下技术方案:一类香豆素衍生物及其制备方法,其特征在于:所述香豆素衍生物的制备方法如下:步骤一:制备e-suberenol(3)
[0006][0007]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物1、双(三叔丁基膦)钯(0)、甲苯、三乙胺、烯烃类化合物2;然后将反应瓶密封并置于90℃油浴中;反应完成后(通过tlc监测,约12分钟),将反应混合物冷却至室温,加入nahco3水溶液,然后搅拌5分钟;用乙酸乙酯转移通过硅胶短柱除去不溶性固体,并用乙酸乙酯洗涤;滤液用乙酸乙酯稀释并用水萃取3遍,饱和氯化钠萃取1遍;合并的有机层用无水na2so4干燥,过滤有机相,浓缩得到粗产物;湿法上样柱层析纯化后,得到化合物e-suberenol:
[0008]
步骤二:(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)乙酸烯丙酯(3-1)的制备:
[0009]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物16-溴-7-甲氧基香豆素、双(三叔丁基膦)钯(0),et3n,乙酸烯丙酯;然后将反应瓶密封并置于100℃油浴中;反应完成后(由tlc监测,约30分钟),将反应混合物冷却至室温,并添加1ml饱和nahco3水溶液,然后搅拌5分钟;然后通过硅藻土过滤反应粗品,并用乙酸乙酯洗涤。用乙酸乙酯稀释滤液,并用水和盐水萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后,获得目标化合物。
[0010]
步骤三:(叔丁基碳酸)(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酐(3-2)的制备:
[0011]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物1、双(三叔
丁基膦)钯(0),甲苯、et3n、丙烯酸(叔丁基碳酸)酐;然后将反应瓶密封并置于100℃油浴中;反应30分钟后,将反应混合物冷却至室温并添加饱和nahco3水溶液,然后搅拌5分钟;然后通过硅藻土过滤反应粗品,并用乙酸乙酯洗涤。用乙酸乙酯稀释滤液,并用水和盐水萃取;合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后,获得目标化合物。
[0012]
步骤四:苄基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酯(3-3)的制备:
[0013]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1、双(三叔丁基膦)钯(0),et3n(82μl,0.6mmol,1.5eq.),丙烯酸苄酯。然后将反应瓶密封并置于100℃油浴中;反应完成后,将反应混合物冷却至室温,并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯洗涤。用乙酸乙酯稀释滤液,并用水和盐水萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后,获得目标化合物。
[0014]
步骤五::(e)-7-甲氧基-6-苯乙烯基-2h-色酮-2-酮(3-4)的制备:
[0015]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1、双(三叔丁基膦)钯(0)、甲苯、et3n、乙烯苯;然后将反应瓶密封并置于100℃油浴中。反应30分钟后,将反应混合物冷却至室温并添加饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯洗涤;用乙酸乙酯稀释滤液,并用水和盐水萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后,获得目标化合物。
[0016]
步骤六:乙基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)-2-甲基丙烯酸酯(3-5)的制备:
[0017]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1、双(三叔丁基膦)钯(0),甲苯、et3n、甲基丙烯酸乙酯;然后将反应瓶密封并置于100℃油浴中。反应30分钟后,将反应混合物冷却至室温并添加饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯洗涤。用乙酸乙酯稀释滤液,并用水和盐水萃取;合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品;通过快速柱色谱纯化后,获得目标化合物。
[0018]
作为本发明的进一步方案,碳酸氢钠饱和溶液可以替换为碳酸氢钾、碳酸钠、碳酸钾、碳酸钙、磷酸一氢钠、磷酸二氢钠、磷酸一氢钾、磷酸二氢钾、磷酸钾、磷酸钠、磷酸钙。
[0019]
作为本发明的进一步方案,所述三叔丁基膦可替换为三苯基膦、三甲基膦、三(邻甲基苯基)磷、三环己基膦、三环己基膦氟硼酸盐、三正丁基磷、4,5-双二苯基膦-9,9-二甲基氧杂蒽、双(2-二苯基磷苯基)醚、三(2-呋喃基)膦、四氟硼酸三叔丁基膦、1,2-双(二苯基膦)乙烷、1,3-双(二苯基膦)丙烷、1,4-双(二苯基膦)丁烷、2-(二叔丁基膦)联苯、2-(二环己基膦基)联苯、2-双环己基膦-2',6'-二甲氧基联苯、2-二环己膦基-2'-(n,n-二甲胺)-联苯、2-二环己基膦-2',4',6'-三异丙基联苯、正丁基二(1-金刚烷基)膦、1,1'-双(二异丙基膦)二茂铁、r-(+)-1,1'-联萘-2.2'-双二苯膦、1.1'-联-2-萘酚、5,5'-双(二苯基磷酰)-4,4'-二-1,3-联苯、双二苯基磷酰联萘、双(2-二苯基磷苯基)醚、1,1-二(二-叔丁基膦基)-二茂铁、2-二叔丁基膦-2',4',6'-三异丙基联苯、四三苯基膦氯化钯、二(三叔丁基膦)钯、[1,1'-双(二叔丁基膦)二茂铁]二氯化钯(ii)或者不添加配体。
[0020]
作为本发明的进一步方案,所述三乙胺可替换为三正丙胺、n,n-二异丙基乙胺、n,n-二乙基苯胺、三正辛胺、n,n-环己基甲胺、吡啶、4-二甲氨基吡啶、1,8-二氮杂双环[5.4.0]十一碳-7-烯、1,4-二氮杂二环[2.2.2]辛烷、四丁基氯化铵、四丁基溴化铵、三乙烯二胺、n-甲基二环己基胺、四丁基氢氧化铵、醋酸钾、醋酸钠、碳酸氢钠、碳酸氢钾、碳酸氢铵、碳酸钠、碳酸钾、碳酸铵、碳酸钙、碳酸铯、磷酸一氢钠、磷酸二氢钠、磷酸一氢钾、磷酸二氢钾、磷酸钾、磷酸钠、磷酸钙或者不添加碱。
[0021]
作为本发明的进一步方案,所述甲苯可替换四氢呋喃、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、苯、二甲苯、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、1,2-二氯乙烷,聚乙二醇、乙腈、氯代苯,二甲基亚砜或者不添加溶剂。
[0022]
作为本发明的进一步方案,所述合成过程中反应温度在40℃-145℃之间。
[0023]
作为本发明的进一步方案,所述溴代底物1为:
[0024][0025]
或其他类似的溴代底物。
[0026]
作为本发明的进一步方案,所述烯烃类化合物2为:
[0027]
或其他烯烃类化合物。
[0028]
与现有技术相比,本发明的有益效果是:
[0029]
1.本专利合成路线以简单易制备的6-溴-7-甲氧基香豆素为起始原料,通过钯催化的heck反应,与烯烃类化合物进行偶联,以较高的产率得到香豆素类衍生物。同时使用不同取代基的溴代底物与2-甲基丁烯-2-醇进行heck反应时,同样以中等到高产率得到烯醇类化合物。本专利路线克服了对环境有危害、操作复杂、产率低等问题,同时避免了高毒性试剂(tbab等)以及高活性碘代物的使用。总体来说,该专利合成路线简捷,原料简单易得,操作简便,催化剂使用量少且廉价易得,衍生物产率较好,不但为香豆素类衍生物的合成提供了一种新方法,也为产品规模化生产及提高生产效率提供了更多可能。
[0030]
2.避免毒性试剂(tbab等)以及难以制备的高活性碘化物的使用;解决了现有技术纯度不高,操作复杂,产率偏低,不能大量生产的问题;提高反应的原子经济性;提供一种工艺稳定,操作简便,合成效率高的烯烃类香豆素类化合物的制备方法。
附图说明
[0031]
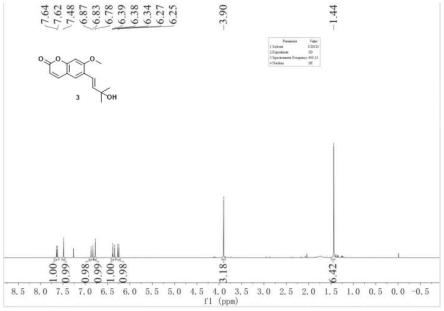
图1为本发明e-suberenol的核磁共振氢谱示意图;
[0032]
图2为本发明e-suberenol的核磁共振碳谱示意图;
[0033]
图3为本发明e-suberenol衍生物1的核磁共振氢谱示意图;
[0034]
图4为本发明e-suberenol衍生物1的核磁共振碳谱示意图;
[0035]
图5为本发明e-suberenol衍生物2的核磁共振氢谱示意图;
[0036]
图6为本发明e-suberenol衍生物2的核磁共振碳谱示意图;
[0037]
图7为本发明e-suberenol衍生物3的核磁共振氢谱示意图;
[0038]
图8为本发明e-suberenol衍生物3的核磁共振碳谱示意图;
[0039]
图9为本发明e-suberenol衍生物4的核磁共振氢谱示意图;
[0040]
图10为本发明e-suberenol衍生物4的核磁共振碳谱示意图;
[0041]
图11为本发明e-suberenol衍生物5的核磁共振氢谱示意图;
[0042]
图12为本发明e-suberenol衍生物5的核磁共振碳谱示意图。
具体实施方式
[0043]
请参阅图1-12,本发明提供一种技术方案:一类香豆素衍生物及其制备方法,所述香豆素衍生物的制备方法如下:
[0044]
步骤一:制备e-suberenol(3)
[0045][0046]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物1(6-溴-7-甲氧基香豆素)6-溴-7-甲氧基香豆素(1.02g,4.0mmol,1.0eq.)、双(三叔丁基膦)钯(0)(164mg,0.032mmol,0.08eq.)、甲苯(15ml)、三乙胺(834μl,6.0mmol,1.5eq.)、烯烃类化合物2(1,1-二甲基烯丙醇)1,1-二甲基烯丙醇(1.88ml,18.0mmol,4.5eq.)。然后将反应瓶密封并置于90℃油浴中。反应完成后(通过tlc监测,约12分钟),将反应混合物冷却至室温,加入nahco3水溶液,然后搅拌5分钟。用乙酸乙酯转移通过硅胶短柱除去不溶性固体,并用乙酸乙酯(100ml)洗涤。滤液用乙酸乙酯稀释并用水萃取3遍,饱和氯化钠萃取1遍。合并的有机层用无水na2so4干燥,过滤有机相,浓缩得到粗产物。湿法上样柱层析(洗脱剂,pe:ea=8:1至2:1)纯化后,得到化合物e-suberenol:(1.02g,98%yield)
[0047]
化合物e-suberenol结构表征数据:
[0048]1h nmr(400mhz,cdcl3)δ7.63(d,1h,j=9.4hz),7.48(s,1h),6.85(d,1h,j=16.2hz),6.78(s,1h),6.36(d,1h,j=16.2hz),6.26(d,1h,j=9.4hz),3.90(s,3h),1.44(s,6h);
[0049]
13
c nmr(100mhz,cdcl3)δ161.20,159.93,155.11,143.49,139.20,125.33,123.78,119.74,113.39,112.20,98.94,71.26,56.00,29.91;
[0050]
ir(kbr):3838.15,3732.92,3433.47,2932.10,2345.14,1728.08,1611.93,1356.06,1210.07,1020.71,830.13,675.92cm-1
;
[0051]
hrms(ei)calcdforc
15h16
o4[m+na]
+
283.0940,found283.0941.
[0052]
步骤二:(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)乙酸烯丙酯(3-1)的制备:
[0053]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物1(6-溴-7-甲氧基香豆素)(100.0mg,0.4mmol,1.0eq.)、双(三叔丁基膦)钯(0)(16.4mg,0.032mmol,0.08eq.),et3n(82μl,0.6mmol,1.5eq.),乙酸烯丙酯(197μl,1.8mmol,4.5eq.)。然后将反应瓶密封并置于100℃油浴中。反应完成后(由tlc监测,约30分钟),将反应混合物冷却至室
温,并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯(30ml)洗涤。用乙酸乙酯(50ml)稀释滤液,并用水(2 10ml)和盐水(10ml)萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后(pe:ea=20:1至2:1),获得目标化合物(62.3mg,收率58%,回收的起始原料,28mg,28%;基于回收的起始原料,收率96%)。
[0054]
(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)乙酸烯丙酯结构数据表征:
[0055]1hnmr(400mhz,cdcl3)δ7.63(d,j=9.4hz,1h),7.50(s,1h),6.91(dt,j=16.2,1.4hz,1h),6.79(s,1h),6.37
–
6.29(m,1h),6.27(d,j=9.4hz,1h),4.74(dd,j=6.5,1.4hz,2h),3.92(s,3h),2.11(s,3h);
[0056]
13
c nmr(100mhz,cdcl3)δ170.85,160.96,160.01,155.49,143.33,127.62,125.95,124.98,123.00,113.57,112.24,99.07,77.23,65.28,56.06,29.40,29.35,21.02;
[0057]
ir(kbr):3377.89,3151.08,1716.08,1627.91,1360.89,1303.77,1234.40,1105.11,980.03,887.18,785.53,561.11,485.83cm-1
.
[0058]
步骤三:(叔丁基碳酸)(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酐(3-2)的制备:
[0059]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管,加入溴代底物1(6-溴-7-甲氧基香豆素)(100.0mg,0.4mmol,1.0eq.)、双(三叔丁基膦)钯(0)(16.4mg,0.032mmol,0.08eq.),甲苯(3ml,在添加之前用氮气鼓泡10分钟)、et3n(82μl,0.6mmol,1.5eq.)、丙烯酸(叔丁基碳酸)酐(256μl,1.8mmol,4.5eq.)。然后将反应瓶密封并置于100℃油浴中。反应30分钟后,将反应混合物冷却至室温并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯(30ml)洗涤。用乙酸乙酯(50ml)稀释滤液,并用水(2 10ml)和盐水(10ml)萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后(pe:ea=20:1至2:1),获得目标化合物(134mg,99%产率)。
[0060]
(叔丁基碳酸)(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酐结构数据表征:
[0061]1h nmr(400mhz,cdcl3)δ7.83(d,j=16.1hz,1h),7.64(d,j=9.5hz,1h),7.58(s,1h),6.82(s,1h),6.45(d,j=16.1hz,1h),6.29(d,j=9.5hz,1h),3.95(s,3h),1.53(s,9h);
[0062]
13
c nmr(100mhz,cdcl3)δ166.35,161.10,160.59,156.60,143.14,137.12,127.80,121.67,121.38,113.89,112.35,99.44,80.63,77.22,56.18,28.21;
[0063]
ir(kbr):3864.28,3616.88,3432.95,33370.99,3279.80,3221.79,2844.13,2778.31,2705.77,2358.27,1719.46,1612.29,1527.40,1202.16,933.12,740.13,593.93,505.01cm-1
.
[0064]
步骤四:苄基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酯(3-3)的制备:
[0065]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1(6-溴-7-甲氧基香豆素)(100.0mg,0.4mmol,1.0eq.)、双(三叔丁基膦)钯(0)(16.4mg,0.032mmol,0.08eq.),et3n(82μl,0.6mmol,1.5eq.),丙烯酸苄酯(277μl,1.8mmol,4.5eq.)。然后将反应瓶密封并置于100℃油浴中。反应完成后(由tlc监测,约30分钟),将反应混合物冷却至室温,并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙
酸乙酯(30ml)洗涤。用乙酸乙酯(50ml)稀释滤液,并用水(2 10ml)和盐水(10ml)萃取;合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后(pe:ea=20:1至2:1),获得目标化合物(108.1,82%产率,回收起始原料,10mg,10%;92%产率,基于回收起始原料)。
[0066]
苄基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)丙烯酸酯结构数据表征:
[0067]1h nmr(400mhz,cdcl3)δ7.95(d,j=16.2hz,1h),7.63(d,j=9.5hz,1h),7.58(s,1h),7.44
–
7.34(m,5h),6.82(s,1h),6.59(d,j=16.1hz,1h),6.29(d,j=9.5hz,1h),5.26(s,2h),3.95(s,3h);
[0068]
13
c nmr(100mhz,cdcl3)δ166.93,161.26,160.54,156.80,143.13,138.91,136.02,128.61,128.34,128.31,121.00,119.35,113.98,112.37,99.53,66.44,56.23;
[0069]
ir(kbr):3051.76,2955.51,1717.38,1618.34,1459.89,1370.05,1278.25,993.34,806.11,685.04,588.96,525.77cm-1
.
[0070]
步骤五:(e)-7-甲氧基-6-苯乙烯基-2h-色酮-2-酮(3-4)的制备:
[0071]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1(6-溴-7-甲氧基香豆素)(100.0mg,0.4mmol,1.0eq.)、双(三叔丁基膦)钯(0)(16.4mg,0.032mmol,0.08eq.)、甲苯(3ml,在添加之前用氮气鼓泡10min)、,et3n(82μl,0.6mmol,1.5eq.),乙烯苯(208μl,1.8mmol,4.5eq.)。然后将反应瓶密封并置于100℃油浴中;反应30分钟后,将反应混合物冷却至室温并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯(30ml)洗涤。用乙酸乙酯(50ml)稀释滤液,并用水(2 10ml)和盐水(10ml)萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,以提供粗产品。通过快速柱色谱纯化后(pe:ea=20:1至2:1),获得目标化合物(101.4mg,93%产率)。
[0072]
(e)-7-甲氧基-6-苯乙烯基-2h-色酮-2-酮结构数据表征:
[0073]1hnmr(400mhz,cdcl3)δ7.69(d,j=9.5hz,1h),7.65(s,1h),7.55(d,j=1.6hz,1h),7.53(d,j=1.6hz,1h),7.41(d,j=12.6hz,1h),7.39
–
7.34(m,2h),7.31
–
7.27(m,1h),7.10(d,j=16.5hz,1h),6.83(s,1h),6.29(d,j=9.5hz,1h),3.96(s,3h);
[0074]
13
c nmr(100mhz,cdcl3)δ161.06,160.07,155.24,143.44,137.36,130.14,128.71,127.82,126.61,124.96,124.28,121.82,113.51,112.37,99.09,77.22,56.09;
[0075]
ir(kbr):3734.51,3065.12,2961.85,1734.40,1616.27,1482.04,1362.57,1109.49,1014.14,873.31,767.40,702.21,486.38cm-1
.
[0076]
步骤六::乙基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)-2-甲基丙烯酸酯(3-5)的制备:
[0077]
在氮气保护下,向双颈圆底烧瓶中加入磁子并加冷凝管、溴代底物1(6-溴-7-甲氧基香豆素)(100.0mg,0.4mmol,1.0eq.)、双(三叔丁基膦)钯(0)(16.4mg,0.032mmol,0.08eq.),甲苯(3ml,在添加之前用氮气鼓泡10分钟)、et3n(82μl,0.6mmol,1.5eq.)、甲基丙烯酸乙酯(226μl,1.8mmol,4.5eq.)。然后将反应瓶密封并置于100℃油浴中。反应30分钟后,将反应混合物冷却至室温并添加1ml饱和nahco3水溶液,然后搅拌5分钟。然后通过硅藻土过滤反应粗品,并用乙酸乙酯(30ml)洗涤。用乙酸乙酯(50ml)稀释滤液,并用水(2 10ml)和盐水(10ml)萃取。合并有机相用无水na2so4干燥,过滤有机相,并在30℃的真空中浓缩,
以提供粗产品。通过快速柱色谱纯化后(pe:ea=20:1至2:1),获得目标化合物:(101.1mg,90%产率)。
[0078]
乙基(e)-3-(7-甲氧基-2-氧代-2h-铬-6-基)-2-甲基丙烯酸酯结构数据表征:
[0079]1h nmr(400mhz,cdcl3)δ7.80
–
7.70(m,1h),7.67(d,j=9.5hz,1h),7.36(s,1h),6.84(s,1h),6.30(d,j=9.5hz,1h),4.29(q,j=7.1hz,2h),3.93(s,3h),2.06(d,j=1.5hz,3h),1.36(t,j=7.1hz,3h);
[0080]
13
c nmr(100mhz,cdcl3)δ168.22,160.89,160.78,155.85,143.34,132.69,129.74,128.92,122.40,113.56,111.84,99.05,61.01,56.19,14.35,14.30;
[0081]
ir(kbr):2976.89,1728.10,1620.70,1490.48,1373.29,1264.36,1115.96,1015.77,910.89,732.44,480.06cm-1
.
[0082]
作为本发明的进一步方案,碳酸氢钠饱和溶液可以替换为碳酸氢钾、碳酸钠、碳酸钾、碳酸钙、磷酸一氢钠、磷酸二氢钠、磷酸一氢钾、磷酸二氢钾、磷酸钾、磷酸钠、磷酸钙。
[0083]
作为本发明的进一步方案,所述三叔丁基膦可替换为三苯基膦、三甲基膦、三(邻甲基苯基)磷、三环己基膦、三环己基膦氟硼酸盐、三正丁基磷、4,5-双二苯基膦-9,9-二甲基氧杂蒽、双(2-二苯基磷苯基)醚、三(2-呋喃基)膦、四氟硼酸三叔丁基膦、1,2-双(二苯基膦)乙烷、1,3-双(二苯基膦)丙烷、1,4-双(二苯基膦)丁烷、2-(二叔丁基膦)联苯、2-(二环己基膦基)联苯、2-双环己基膦-2',6'-二甲氧基联苯、2-二环己膦基-2'-(n,n-二甲胺)-联苯、2-二环己基膦-2',4',6'-三异丙基联苯、正丁基二(1-金刚烷基)膦、1,1'-双(二异丙基膦)二茂铁、r-(+)-1,1'-联萘-2.2'-双二苯膦、1.1'-联-2-萘酚、5,5'-双(二苯基磷酰)-4,4'-二-1,3-联苯、双二苯基磷酰联萘、双(2-二苯基磷苯基)醚、1,1-二(二-叔丁基膦基)-二茂铁、2-二叔丁基膦-2',4',6'-三异丙基联苯、四三苯基膦氯化钯、二(三叔丁基膦)钯、[1,1'-双(二叔丁基膦)二茂铁]二氯化钯(ii)或者不添加配体。
[0084]
作为本发明的进一步方案,所述三乙胺可替换为三正丙胺、n,n-二异丙基乙胺、n,n-二乙基苯胺、三正辛胺、n,n-环己基甲胺、吡啶、4-二甲氨基吡啶、1,8-二氮杂双环[5.4.0]十一碳-7-烯、1,4-二氮杂二环[2.2.2]辛烷、四丁基氯化铵、四丁基溴化铵、三乙烯二胺、n-甲基二环己基胺、四丁基氢氧化铵、醋酸钾、醋酸钠、碳酸氢钠、碳酸氢钾、碳酸氢铵、碳酸钠、碳酸钾、碳酸铵、碳酸钙、碳酸铯、磷酸一氢钠、磷酸二氢钠、磷酸一氢钾、磷酸二氢钾、磷酸钾、磷酸钠、磷酸钙或者不添加碱。
[0085]
作为本发明的进一步方案,所述甲苯可替换四氢呋喃、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮、苯、二甲苯、1,4-二氧六环、乙二醇二甲醚、乙二醇二乙醚、1,2-二氯乙烷,聚乙二醇、乙腈、氯代苯,二甲基亚砜或者不添加溶剂。
[0086]
作为本发明的进一步方案,所述合成过程中反应温度在40℃-145℃之间
[0087]
作为本发明的进一步方案,溴代底物为:
[0088][0089]
或其他类似的溴代底物。
[0090]
作为本发明的进一步方案,烯烃类化合物为:
[0091]
或其他烯烃类化合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1