一种Belzutifan的中间体的制备方法
一种belzutifan的中间体的制备方法
技术领域
1.本发明属于制药技术领域,具体涉及一种belzutifan的中间体的制备方法。
背景技术:
2.希佩尔
·
林道综合征(von hippel-lindauvon hippel-lindau,vhl)是一种罕见的常染色体显性遗传病,通常由vhl胚系突变引起。vhl作为e3泛素连接酶,可使缺氧诱导因子hif-α亚基泛素化,从而导致hif发生泛素化降解。因vhl基因突变,导致hif-2α在vhl疾病相关的肾细胞癌中过表达。hif-2α抑制剂可阻断hif近端大部分信号通路,抑制肾透明细胞癌肿瘤细胞的生长。2021年8月,belzutifan获fda批准用于无需立即手术的、与vhl综合征相关的肾细胞癌、中枢神经系统血管细胞瘤以及胰腺神经内分泌肿瘤。
3.迄今为止,belzutifan是唯一获批的vhl相关肿瘤全身治疗药物。目前报道的belzutifan的工业化合成路线如路线1,其中化合物3-氟-5-((7-(甲磺酰基)-2,3-二氢螺[茚-1,2'-[1,3]二氧戊环]-4-基)氧基)苯甲腈是其关键中间体,也是合成的技术难点和制约产品成本控制的关键所在。现有技术中,该中间体的合成方法如下:
[0004]
方法一:文献org.process res.dev.,2022,26,508-515的合成方法(如路线1所示)。该方法以二氢香豆素(a1)为起始原料,与nbs在一个当量的路易斯酸存在下,发生溴代反应,生成中间体b1。该反应会产生1%左右的二溴取代的副产物(b1’),反应放热严重,需要在低温下缓慢加入nbs。中间体b1与3,5-二氟苯甲腈在碳酸钾作用下,发生snar反应生成中间体c1。反应中碱促使中间体b1开环形成c1
‘
,c1
‘
与3,5-二氟苯甲腈形成双-snar加成的副产物c1“。为了降低的副产物的含量,体系中需要加入0.3当量的18-冠醚-6,增加了合成的成本。中间体c1随后发生傅克酰基化反应生成中间体d1。中间体d1与甲基亚磺酸钠发生取代反应。最后乙二醇保护,生成重要的缩醛中间体vi。
[0005]
[0006][0007]
方法二:文献j.med.chem.,2018,61,9691-9721、文献j.med.chem.,2019,62,6876-6893及专利wo2015035223a1的合成方法(如路线2所示)。该方法以对氟苯酚(a2)为原料,首先发生取代反应得到中间体b2,随后发生重排和傅克反应得到中间体c2。中间体c2与二甲基硫代甲酰氯发生缩合反应,后经高温重排、水解生成中间体f2。中间体f2与碘甲烷反应,后被oxone氧化剂氧化,生成中间体h2。中间体h2与乙二醇反应,生成缩醛中间体i2,后采用碳酸氢铯做碱,与3-氟-5-羟基苯甲腈反应生成重要中间体vi。通过分析以上合成路线,我们发现路线存在很多明显缺陷:路线较长,共经历9步反应才最终得到终产品。合成中间体c2时,因为要发生重排反应,所以采用的是无溶剂反应,需要将氯化铝加热到融化温度,180℃反应,而且反应后,体系发生碳化现象明显,很难进行后处理。路线中甲磺酰基的构建共经历5步反应,原子经济性差,且引入使用了有基因毒性的碘甲烷试剂,合成试剂危险性大,不适宜工业化生产。
[0008][0009]
方法三:专利wo2016145236a1的合成方法(如路线3所示)。该方法以4-(甲硫基)苯酚(a3)为原料,这个原料的制备要使用恶臭的试剂二甲基二硫化物。原料a3与过量的多聚甲醛反应,生成中间体b3。中间体b3和丙二酸(亚)异丙酯在磷酸钾作用下,获得中间体c3。中间体c3被还原生成中间体d3。中间体d3与3,5-二氟苯甲腈发生snar反应生成中间体e3。但该反应中会产生双-snar加成的副产物(e3’),为避免该副产物的产生,需要5当量的3,5-二氟苯甲腈,但最终仍会产生3%左右。中间体e3随后发生傅克酰基化反应生成中间体g3。中间体g3发生氧化反应,生成甲磺酰基中间体h3,最后与乙二醇反应,生成重要的缩醛中间体vi。整体看,该方法7步反应,收率大约20%。反应中3-氟-5-羟基苯甲腈使用5个当量,显著增加合成成本。该路线使用多聚甲醛和双氧水等高危险试剂,不适宜工业化生产。
[0010][0011]
因此,亟需一种产率高、纯度高、成本低、安全性高、反应路线短的3-氟-5-((7-(甲磺酰基)-2,3-二氢螺[茚-1,2'-[1,3]二氧戊环]-4-基)氧基)苯甲腈的制备方法。
技术实现要素:
[0012]
为解决上述技术问题,本发明提供以下技术方案。
[0013]
一种式vi所示化合物的制备方法,其包括:
[0014][0015]
式v所示化合物在碳酸钾存在的条件下,在溶剂1中与3-氟-5-羟基苯甲腈发生第一反应,得到式vi所示化合物。
[0016]
在一些实施例中,所述溶剂1包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、乙腈或n-甲基吡咯烷酮中的至少一种。
[0017]
在一些实施例中,所述式v所示化合物与3-氟-5-羟基苯甲腈的投料摩尔比为1.0:1.0~1.0:1.3。在一些实施例中,所述式v所示化合物与3-氟-5-羟基苯甲腈的投料摩尔比为1.0:1.0、1.0:1.1、1.0:1.2或1.0:1.3。
[0018]
在一些实施例中,所述式v所示化合物与碳酸钾的投料摩尔比为1.0:1.5~1.0:3.0。在一些实施例中,所述式v所示化合物与碳酸钾的投料摩尔比为1.0:1.5、1.0:2.0、1.0:2.5或1.0:3.0。
[0019]
在一些实施例中,所述第一反应的反应温度为25℃~120℃。在一些实施例中,所
述第一反应的反应温度为50℃~100℃。在一些实施例中,所述第一反应的反应温度为70℃~95℃。在一些实施例中,所述第一反应的反应温度为25℃、30℃、35℃、40℃、45℃、50℃、55℃、60℃、65℃、70℃、75℃、80℃、85℃、90℃、95℃、100℃、105℃、110℃、115℃或120℃。
[0020]
在一些实施例中,所述式v所示化合物的制备方法包括:
[0021][0022]
式iv所示化合物在溶剂2中与乙二醇和对甲基苯磺酸或其水合物发生第二反应,得到式v所示化合物。
[0023]
在一些实施例中,所述溶剂2包括苯或甲苯中的至少一种。
[0024]
在一些实施例中,所述式iv所示化合物与乙二醇的投料摩尔比为1.0:3.0~1.0:10.0。在一些实施例中,所述式iv所示化合物与乙二醇的投料摩尔比为1.0:3.0、1.0:4.0、1.0:5.0、1.0:6.0、1.0:7.0、1.0:8.0、1.0:9.0或1.0:10.0。
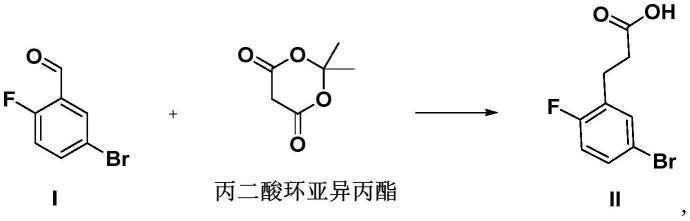
[0025]
在一些实施例中,所述式iv所示化合物与对甲基苯磺酸或其水合物的投料摩尔比为1.000:0.003~1.0:0.020。在一些实施例中,所述式iv所示化合物与对甲基苯磺酸或其水合物的投料摩尔比为1.000:0.010~1.0:0.020。在一些实施例中,所述式iv所示化合物与对甲基苯磺酸或其水合物的投料摩尔比为1.000:0.003、1.000:0.005、1.000:0.010、1.000:0.015或1.000:0.020。
[0026]
在一些实施例中,所述第二反应的反应温度为120℃~140℃。在一些实施例中,所述第二反应的反应温度为120℃、125℃、130℃、135℃或140℃。
[0027]
在一些实施例中,所述式iv所示化合物的制备方法包括:
[0028][0029]
惰性气体氛围下,式iii所示化合物在催化剂a存在的条件下,在溶剂3中与甲基亚磺酸钠发生第三反应,得到式iv所示化合物。
[0030]
在一些实施例中,所述溶剂3包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、甲苯或n-甲基吡咯烷酮中的至少一种。
[0031]
在一些实施例中,所述催化剂a包括碘化亚铜或溴化亚铜中的至少一种。
[0032]
在一些实施例中,所述式iii所示化合物与甲基亚磺酸钠的投料摩尔比为1.0:1.0~1.0:2.0。在一些实施例中,所述式iii所示化合物与甲基亚磺酸钠的投料摩尔比为1.0:1.0、1.0:1.1、1.0:1.2、1.0:1.3、1.0:1.4、1.0:1.5、1.0:1.6、1.0:1.7、1.0:1.8、1.0:1.9或1.0:2.0。
[0033]
在一些实施例中,所述式iii所示化合物与催化剂a的投料摩尔比为1.0:2.0~1.0:8.0。在一些实施例中,所述式iii所示化合物与催化剂a的投料摩尔比为1.0:3.0~
1.0:8.0。在一些实施例中,所述式iii所示化合物与催化剂a的投料摩尔比为1.0:2.0、1.0:3.0、1.0:4.0、1.0:5.0、1.0:6.0、1.0:7.0或1.0:8.0。
[0034]
在一些实施例中,所述第三反应的反应温度为80℃~120℃。在一些实施例中,所述第三反应的反应温度为90℃-110℃。在一些实施例中,所述第三反应的反应温度为80℃、85℃、90℃、95℃、100℃、105℃、110℃、115℃或120℃。
[0035]
在一些实施例中,所述惰性气体为氦气、氖气、氩气、氪气或氮气中的至少一种。
[0036]
在一些实施例中,所述式iii所示化合物的制备方法包括:
[0037][0038]
式ii所示化合物在有机溶剂中与酰氯化试剂发生酰氯化反应,将式ii所示化合物酰氯化,再加入催化剂b,发生傅克酰基化反应,得到式iii所示化合物。
[0039]
在一些实施例中,所述酰氯化试剂包括二氯亚砜、草酰氯、三氯化磷、五氯化磷中的至少一种。
[0040]
在一些实施例中,所述催化剂b包括三氯化铝、三氯化铁、四氯化锡、三氟化硼、四氯化锑、氯化锌中的至少一种。
[0041]
在一些实施例中,所述有机溶剂包括二氯甲烷、氯仿、1,4-二氧六环中的至少一种。
[0042]
在一些实施例中,所述式ii所示化合物与酰氯化试剂的投料摩尔比为1.0:1.0~1.0:2.0。在一些实施例中,所述式ii所示化合物与酰氯化试剂的投料摩尔比为1.0:1.2~1.0:2.0。在一些实施例中,所述式ii所示化合物与酰氯化试剂的投料摩尔比为1.0:1.0、1.0:1.1、1.0:1.2、1.0:1.3、1.0:1.4、1.0:1.5、1.0:1.6、1.0:1.7、1.0:1.8、1.0:1.9或1.0:2.0。
[0043]
在一些实施例中,所述式ii所示化合物与催化剂b的投料摩尔比为1.0:1.0~1.0:5.0。在一些实施例中,所述式ii所示化合物与催化剂b的投料摩尔比为1.0:2.0~1.0:5.0。在一些实施例中,所述式ii所示化合物与催化剂b的投料摩尔比为1.0:1.0、1.0:2.0、1.0:3.0、1.0:4.0或1.0:5.0。
[0044]
在一些实施例中,所述酰氯化反应还可以包括加入n,n-二甲基甲酰胺;用以促进酰氯化反应的进行。
[0045]
在一些实施例中,所述酰氯化反应中,每1mol所述式ii所示化合物对应投料n,n-二甲基甲酰胺0~0.5ml。在一些实施例中,所述酰氯化反应中,每1mol所述式ii所示化合物对应投料n,n-二甲基甲酰胺0.1ml~0.5ml。在一些实施例中,所述酰氯化反应中,每1mol所述式ii所示化合物对应投料n,n-二甲基甲酰胺0.2ml~0.5ml。
[0046]
在一些实施例中,所述式ii所示化合物的制备方法包括:
[0047][0048]
丙二酸环亚异丙酯在甲酸和三乙胺存在的条件下,在溶剂4中与式i所示化合物反应,得到式ii所示化合物。
[0049]
在一些实施例中,所述溶剂4包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、1,4-二氧六环、甲苯或n-甲基吡咯烷酮中的至少一种。
[0050]
在一些实施例中,式i所示化合物与丙二酸环亚异丙酯的投料摩尔比为1.0:1.0~1.0:1.5。在一些实施例中,式i所示化合物与丙二酸环亚异丙酯的投料摩尔比为1.0:1.0、1.0:1.1、1.0:1.2、1.0:1.3、1.0:1.4或1.0:1.5。
[0051]
在一些实施例中,式i所示化合物与三乙胺的投料摩尔比为1.0:1.2~1.0:2.0。在一些实施例中,式i所示化合物与三乙胺的投料摩尔比为1.0:1.5~1.0:2.0。在一些实施例中,式i所示化合物与三乙胺的投料摩尔比为1.0:1.2、1.0:1.3、1.0:1.4、1.0:1.5、1.0:1.6、1.0:1.7、1.0:1.8、1.0:1.9或1.0:2.0。
[0052]
在一些实施例中,式i所示化合物与甲酸的投料摩尔比为1.0:2.0~1.0:5.0。在一些实施例中,式i所示化合物与甲酸的投料摩尔比为1.0:3.0~1.0:5.0。在一些实施例中,式i所示化合物与甲酸的投料摩尔比为1.0:2.0、1.0:2.5、1.0:3.0、1.0:3.5、1.0:4.0、1.0:4.5或1.0:5.0。
[0053]
在一些实施例中,所述式ii所示化合物的制备方法的反应温度为80℃~150℃。在一些实施例中,所述式ii所示化合物的制备方法的反应温度为90℃~120℃。在一些实施例中,所述式ii所示化合物的制备方法的反应温度为80℃、85℃、90℃、95℃、100℃、105℃、110℃、120℃、130℃、140℃或150℃。
[0054]
有益效果
[0055]
相比现有技术,本发明的某一实施例具有以下至少一种有益效果:
[0056]
(1)式ii所示化合物的制备方法中,相比其他反应物和其他还原试剂,本发明采用丙二酸环亚异丙酯作为反应物,采用甲酸和三乙胺作为还原试剂,更有利于提高式ii所示化合物的产率,且不需要加入例如镁等易燃易爆的危险试剂,安全环保,成本低,反应路线短,操作简单。
[0057]
(2)式iv所示化合物的制备方法中,相比其他催化剂,本发明采用碘化亚铜或溴化亚铜作为催化剂,更有利于提高式iv所示化合物的产率。
[0058]
(3)式vi所示化合物的制备方法中,相比其他碱,本发明采用碳酸钾,更有利于提高式vi所示化合物的产率。
[0059]
(4)与背景技术方法一相比,本发明采用的技术路线,副产物少,在最后才引入第二个关键片段,可大大节约原料成本。
[0060]
(5)本发明提供的全部工艺路线为5步,总收率高达47%。与背景技术的方法二和方法三相比,缩短了反应路线,避免了危险试剂的使用。综合来看,本发明提供的合成路线
更加合理,反应步骤简洁,原料廉价易得,易于工业化生产。
[0061]
术语说明
[0062]
除非另外说明,否则如本文使用的以下术语和短语意图具有以下含义:
[0063]
本发明中,如“化合物i”和“式i所示化合物”的表述,表示的是同一个化合物。
[0064]
本发明中“室温”指的是环境温度,温度由大约10℃到大约40℃。在一些实施例中,“室温”指的是温度由大约20℃到大约30℃;在另一些实施例中,“室温”指的是温度由大约25℃到大约30℃;在又一些实施例中,“室温”指的是10℃、15℃、20℃、25℃、30℃、35℃、40℃等。
[0065]
术语“m”表示mol/l。
[0066]
术语“催化剂a”、“催化剂b”中的“a”和“b”仅为了区别不同反应中的催化剂,并无其他实际意义,仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量;由此,“催化剂a”、“催化剂b”特征可以明示或者隐含地包括一个或者更多个该特征。
[0067]
术语“溶剂1”、“溶剂2”、“溶剂3”、“溶剂4”中的“1”、“2”、“3”、“4”仅为了区别不同反应中的溶剂,仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量;由此,“溶剂1”、“溶剂2”、“溶剂3”、“溶剂4”中的“1”、“2”、“3”、“4”特征可以明示或者隐含地包括一个或者更多个该特征。
[0068]
术语“第一反应”、“第二反应”、“第三反应”中的“第一”、“第二”、“第三”、“第四”仅为了区别不同反应,仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。
[0069]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
具体实施例
[0070]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例以对本发明作进一步的详细说明。
[0071]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0072]
实施例1:式ii所示化合物的制备
[0073][0074]
取甲酸(40.8g,886.6mmol)于0℃与三乙胺(44.9g,443.3mmol)混合,再与丙二酸环亚异丙酯(51.1g,354.6mmol)混合,然后与5-溴-2-氟苯甲醛(60g,295.5mmol)的n,n-二甲基甲酰胺(200ml)溶液混合,室温搅拌1h后升温至100℃,继续反应5h,冷却,水洗,乙酸乙酯萃取三次,合并有机层,饱和氯化钠水溶液洗一遍,无水硫酸钠干燥,旋去有机溶液,用乙酸乙酯:石油醚(v/v)=1:3的混合溶液重结晶,得式ii所示化合物(66g,90%)。取所得式ii所示化合物,检测氢谱,结果如下:
[0075]1h nmr(400mhz,cdcl3):δ7.34
–
7.30(m,2h,),6.92(m,1h),3.07
–
2.90(m,2h),2.73
–
2.68(m,2h).
[0076]
实施例2:式iii所示化合物的制备
[0077][0078]
将式ii所示化合物(66g,267.1mmol)溶于二氯甲烷(300ml)中,于0℃与二氯亚砜(38.2g,320.52mmol)混合,再加入2滴(约0.1ml)dmf(n,n-二甲基甲酰胺),室温反应2h;减压浓缩,再用二氯甲烷(500ml)复溶,于0℃,与氯化铝(71.2g,534.2mmol)混合。温度升至室温继续反应20h;反应体系冷却至0℃,缓慢倾入到500ml冰水中;用硅藻土抽滤,用二氯甲烷反复洗滤饼,最后将滤液合并,分液,将有机层用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,旋去有机溶液,柱层析,得式iii所示化合物(47g,77%)。取所得式iii所示化合物,检测氢谱,结果如下:
[0079]1h nmr(600mhz,cdcl3)δ7.51
–
7.49(m,1h),7.15
–
7.12(m,1h),3.12
–
3.10(m,2h),2.79
–
2.77(m,2h).
[0080]
实施例3:式iv所示化合物的制备
[0081][0082]
将式iii所示化合物(47g,205.2mmol)与二甲基亚砜(300ml)混合,再与碘化亚铜(117g,615.6mmol)和甲基亚磺酸钠(25.2g,246.2mmol)混合。氩气保护下,100℃反应24h。冷却,水洗,乙酸乙酯萃取三次,合并有机层,饱和氯化钠水溶液洗涤,无水硫酸钠干燥,旋
去有机溶液,柱层析得式iv所示化合物(39g,83%)。取所得式iv所示化合物,检测氢谱,结果如下:
[0083]1h nmr(400mhz,cdcl3)δ8.18
–
8.14(m,1h),7.42(t,j=8.1hz,1h),3.42(s,3h),3.28
–
3.19(m,2h),2.90
–
2.83(m,2h).
[0084]
实施例4:式v所示化合物的制备
[0085][0086]
将式iv所示化合物(39g,170.9mmol)溶于甲苯(300ml)中,分别加入乙二醇(53g,854.5mmol)和对甲基苯磺酸一水合物(323mg,1.7mmol),加热至130℃,分水器分水,反应24h。减压除去甲苯,饱和碳酸氢钠水溶液(50ml)洗一遍,乙酸乙酯萃取两次,合并有机层,饱和氯化钠水溶液洗一遍,无水硫酸钠干燥,旋去有机溶液,得式v所示化合物(44g,95%)。取所得式v所示化合物,检测氢谱,结果如下:
[0087]1h nmr(400mhz,cdcl3)δ7.96
–
7.92(m,1h),7.12
–
7.08(m,1h),4.45
–
4.23(m,2h),4.17
–
3.88(m,2h),3.18(s,3h),2.91(t,j=6.8hz,2h),2.22(t,j=6.8hz,2h).
[0088]
实施例5:式vi所示化合物的制备
[0089][0090]
将式v所示化合物(44g,161.6mmol)与n,n-二甲基甲酰胺(300ml)混合,再与3-氟-5-羟基苯甲腈(24.4g,177.7mmol)和碳酸钾(44.6g,323.2mmol)混合,加热至90℃,反应5h。降至室温,水洗,乙酸乙酯萃取三次,合并有机层,饱和氯化钠水溶液洗一遍,无水硫酸钠干燥,旋去有机溶液,柱层析得式vi所示化合物(54g,86%)。取所得式vi所示化合物,检测氢谱,结果如下:
[0091]1h nmr(400mhz,cdcl3)δ8.03(d,j=8.5hz,1h),7.19
–
7.16(m,1h),7.10
–
7.09(m,1h),7.02(d,j=8.5hz,1h),6.99
–
6.96(m,1h),4.45
–
4.35(m,2h),4.14
–
4.04(m,2h),3.30(s,3h),2.88(t,j=6.8hz,2h),2.29(t,j=6.8hz,3h).
[0092]
对比例1:式vi所示化合物的制备
[0093][0094]
将式v所示化合物(44g,161.6mmol)与n,n-二甲基甲酰胺(300ml)混合,再与3-氟-5-羟基苯甲腈(24.4g,177.7mmol)和碳酸铯(105.3g,323.2mmol)混合,加热至90℃,反应5h。降至室温,水洗,乙酸乙酯萃取三次,合并有机层,饱和氯化钠水溶液洗一遍,无水硫酸钠干燥,旋去有机溶液,柱层析得式vi所示化合物(13.8g,22%)。
[0095]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1