一种(S)-2-(4-溴苯基)丙酸的制备方法与流程
一种(s)-2-(4-溴苯基)丙酸的制备方法
技术领域
1.本发明涉及生物制药合成技术领域,特别是涉及一种(s)-2-(4-溴苯基)丙酸的制备方法。
背景技术:
2.(s)-2-(4-溴苯基)丙酸作为重要的化学中间体,广泛应用于各种医药合成(如细胞周期蛋白依赖性激酶cdk 12/13或组织蛋白酶k抑制剂)。(s)-2-(4-溴苯基)丙酸的结构为:
[0003][0004]
目前,现有技术中,(s)-2-(4-溴苯基)丙酸的制备方法主要有以下四种:
[0005]
(1)通过金属催化剂(如pd,ni),手性膦配体不对称合成单一的手性异构体,但有重金属污染,且对映选择性不高(85%~91%e.e.)。
[0006]
(2)通过马肝醇脱氢酶催化醛的歧化反应,但使用本方法,反应体积较大,不适宜反应的放大生产,且对映选择性过量值只有84%。
[0007]
(3)如申请号为cn202080026783.4,名称为“作为选择性cdk12/13抑制剂的取代5-环丙基-1h-吡唑-3-基-胺衍生物”的发明专利,通过手性恶唑烷酮(evans辅基)或手性醇诱导合成单一构型酰胺或酯,但本方法在产物脱除时需用到危险试剂过氧化氢,容易对实验室安全或生产安全造成较大威胁。
[0008]
(4)如申请号为us7312353,名称为“夹心半胱氨酸蛋白酶抑制剂”的美国发明专利中,使用化学拆分法,该方法需先和手性胺反应合成盐,然后在通过碱化游离方,最后得到单一构型,但该方法的整体反应周期长,且可能需要多次拆分,才可得到合格的手性异构体。
[0009]
因此,本领域需要一种(s)-2-(4-溴苯基)丙酸的制备方法,在保持较高的对映选择性的前提下能够适应工业放大生产,还能保证反应的安全,在减少周期、降低成本的同时,做到绿色环保。
技术实现要素:
[0010]
为解决上述技术问题,本发明提供以下技术方案:
[0011]
一种(s)-2-(4-溴苯基)丙酸的制备方法,在有机溶剂和缓冲溶液存在下,底物在水解酶的催化下,发生不对称水解反应,生成产物(s)-2-(4-溴苯基)丙酸,所述制备方法的步骤如下:
[0012]
s1、将有机溶剂、缓冲溶液、底物和水解酶加入反应器中,调节反应体系ph值并使其保持在6.0-8.0,在20℃-35℃条件下,进行不对称水解反应,反应时间为20-33小时;
[0013]
s2、调节ph值至9,对反应液进行过滤并萃取,再调节水相的ph至3后再次进行萃
取,合并有机相后进行除水和浓缩,获得粗产物;
[0014]
s3、将获得的粗产物进行打浆、过滤和干燥,得到纯产物;
[0015]
s4、回收反应液中的(r)-2-(4-溴苯基)丙酸甲酯,对(r)-2-(4-溴苯基)丙酸甲酯进行消旋化;
[0016]
s5、将消旋化后的(r)-2-(4-溴苯基)丙酸甲酯作为底物,重复s1、s2和s3的操作,获得纯产物(s)-2-(4-溴苯基)丙酸。
[0017]
具体地,所述底物选自2-(4-溴苯基)丙酸甲酯或其酯类衍生物。
[0018]
具体地,所述水解酶选自脂肪酶、酯酶或蛋白酶。
[0019]
具体地,所述有机溶剂选自二甲基亚砜、二甲基甲酰胺、乙腈、甲醇、乙醇、异丙醇、1,4-二氧六环、四氢呋喃、异丙醚、甲叔醚、正己烷、正庚烷中的一种或多种的混合。
[0020]
优选地,所述有机溶剂选自异丙醚、正己烷、正庚烷中的一种或多种的混合。
[0021]
具体地,所述缓冲溶液为可在ph6.0-8.0范围内缓冲的缓冲液。
[0022]
优选地,所述缓冲液为磷酸盐缓冲溶液。
[0023]
具体地,所述磷酸盐缓冲溶液的摩尔浓度为0.1-0.3mol/l,ph值为6.0-8.0。
[0024]
具体地,所述不对称水解反应的ph值采用氢氧化钠溶液进行调节,所述氢氧化钠溶液的浓度为0.5mol/l或1.0mol/l。
[0025]
具体地,所述s2中,所述过滤是指使用硅藻土过滤反应液,去除酶蛋白,所述萃取的萃取剂为乙酸乙酯,所述除水的干燥剂的为无水硫酸钠。
[0026]
具体地,所述s3中,所述打浆的溶剂为甲叔醚,所述甲叔醚的体积为粗产物的0.5倍。
[0027]
本发明的有益效果包括:
[0028]
(1)本发明反应工艺设计合理,使用水解酶催化的方法高效制备(s)-2-(4-溴苯基)丙酸,相比起现有技术,本发明提供的制备方法,由外消旋体一步制备单一构型的羧酸,实现了较高底物浓度(100g/l)和较大反应规模(大于50kg),较高对映选择性得到(s)-2-(4-溴苯基)丙酸。
[0029]
(2)本发明提供的反应方法,通过回收(r)-2-(4-溴苯基)丙酸甲酯,将其消旋化,再次酶催化水解,将反应总收率提高至68%,避免了化学原料的浪费。除此之外,本发明提供的方法,原料廉价易得,因此也降低了反应成本。
[0030]
(3)与不对称合成法相比,本发明提供的制备方法反应条件温和,绿色环保,避免了使用危险试剂h2o2及金属催化剂,保证了实验室或生产中的安全性。
[0031]
(4)与化学拆分法相比,本发明提供的制备方法,产物的收率显著提高,且缩短了反应周期。
[0032]
(5)与马肝醇脱氢酶催化醛的歧化反应相比,本发明的制备方法可适应的反应底物浓度更大,更适宜工业放大生产,实用性显著提升。
附图说明
[0033]
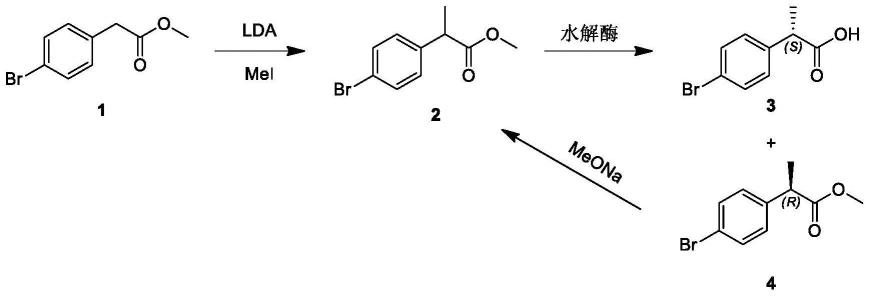
图1为本发明的(s)-2-(4-溴苯基)丙酸的合成路线图;
[0034]
图中,lda为二异丙基氨基锂,mei为碘甲烷,meona为甲醇钠。
具体实施方式
[0035]
下面将对本发明的技术方案进行清楚、完整的描述,显然,所描述的实施例是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0036]
在下述所有实施例中,相关化学试剂英文缩写与中文名称对应关系如下:
[0037]
thf为四氢呋喃,lda为二异丙基氨基锂,mei为碘甲烷,naoh为氢氧化钠。
[0038]
实施例1反应底物2-(4-溴苯基)丙酸甲酯的合成
[0039]
本发明提供的制备方法,反应底物2-(4-溴苯基)丙酸甲酯的合成过程如下:
[0040][0041]
本发明中底物2-(4-溴苯基)丙酸甲酯的获得方法为:将thf(232l)加入到1000l釜中,将化合物1(58kg)加入到釜中。冷却至-78度,在氮气保护下,将lda(2.00m,143l)滴加入到1000l釜中。滴加完毕,在-60~-78℃反应0.5小时。将mei(40.9kg)滴加到反应体系中。滴加完毕,升温至25℃,并在此温度继续反应12小时。加60l氯化铵水溶液淬灭反应,维持温度小于25℃,继续搅拌0.5小时。使用乙酸乙酯(60l*2)萃取,有机相合并,旋干得到粗品化合物2(58kg),直接用于下一步产物(s)-2-(4-溴苯基)丙酸的合成。
[0042]
实施例2以正己烷作为有机溶剂合成产物(s)-2-(4-溴苯基)丙酸
[0043]
如图1所示的合成路线,在一干净250ml玻璃夹套反应瓶中加入80ml 0.1mol/l磷酸盐缓冲液,称取脂肪酶(0.5g)加入到磷酸盐缓冲液中搅拌均匀。将反应体系控温在30℃。滴加底物(10g)的正己烷(20ml)溶液,开始反应。反应过程中,利用0.5mol/l naoh溶液控制ph在7左右。20小时后终止反应,将反应ph调至9,将反应液用硅藻土过滤,滤饼用乙酸乙酯淋洗,滤液用乙酸乙酯萃取。然后将水相调至ph 3,用乙酸乙酯萃取,将调酸后所得有机相合并,加入无水硫酸钠除水,再过滤获得有机相,减压浓缩得到粗品(s)-2-(4-溴苯基)丙酸4.9g。然后将粗品用0.5倍体积甲叔醚打浆过滤,滤饼干燥得到纯品(s)-2-(4-溴苯基)丙酸4.7g。
[0044]
实施例3以正庚烷作为有机溶剂合成产物(s)-2-(4-溴苯基)丙酸
[0045]
如图1所示的合成路线,在一干净250ml玻璃夹套反应瓶中加入80ml 0.1mol/l磷酸盐缓冲液,称取脂肪酶(0.5g)加入到磷酸盐缓冲液中搅拌均匀。将反应体系控温在30℃。滴加底物(10g)的正庚烷(20ml)溶液,开始反应。反应过程中,利用0.5mol/l naoh溶液控制ph在7左右。22小时后终止反应,将反应ph调至9,将反应液用硅藻土过滤,滤饼用乙酸乙酯淋洗,滤液用乙酸乙酯萃取。然后将水相调至ph 3,用乙酸乙酯萃取,将调酸后所得有机相合并,加入无水硫酸钠除水,再过滤获得有机相,减压浓缩得到粗品(s)-2-(4-溴苯基)丙酸4.85g。然后将粗品用0.5倍体积甲叔醚打浆过滤,滤饼干燥得到纯品(s)-2-(4-溴苯基)丙酸4.75g。
[0046]
实施例4以异丙醚作为有机溶剂合成产物(s)-2-(4-溴苯基)丙酸
[0047]
如图1所示的合成路线,在一干净250ml玻璃夹套反应瓶中加入80ml 0.1mol/l磷酸盐缓冲液,称取脂肪酶(0.5g)加入到磷酸盐缓冲液中搅拌均匀。将反应体系控温在30℃。
滴加底物(10g)的异丙醚(20ml)溶液,开始反应。反应过程中,利用0.5mol/l naoh溶液控制ph在7左右。30小时后终止反应,将反应ph调至9,将反应液用硅藻土过滤,滤饼用乙酸乙酯淋洗,滤液用乙酸乙酯萃取。然后将水相调至ph 3,用乙酸乙酯萃取,将调酸后所得有机相合并,加入无水硫酸钠除水,再过滤获得有机相,减压浓缩得到粗品(s)-2-(4-溴苯基)丙酸4.7g。然后将粗品用0.5倍体积甲叔醚打浆过滤,滤饼干燥得到纯品(s)-2-(4-溴苯基)丙酸4.5g。
[0048]
实施例5通过调整ph值以缩短(s)-2-(4-溴苯基)丙酸的合成周期
[0049]
如图1所示的合成路线,在一干净250ml玻璃夹套反应瓶中加入80ml 0.1mol/l磷酸盐缓冲液,称取脂肪酶(0.5g)加入到磷酸盐缓冲液中搅拌均匀。将反应体系控温在30℃。滴加底物(10g)的正己烷(20ml)溶液,开始反应。反应过程中,利用0.5mol/l naoh溶液控制ph在6左右。20小时后终止反应,将反应ph调至9,将反应液用硅藻土过滤,滤饼用乙酸乙酯淋洗,滤液用乙酸乙酯萃取。然后将水相调至ph 3,用乙酸乙酯萃取,将调酸后所得有机相合并,加入无水硫酸钠除水,再过滤获得有机相,减压浓缩得到粗品(s)-2-(4-溴苯基)丙酸4.6g。然后将粗品用0.5倍体积甲叔醚打浆过滤,滤饼干燥得到纯品(s)-2-(4-溴苯基)丙酸4.4g。
[0050]
实施例6通过调整ph值以缩短(s)-2-(4-溴苯基)丙酸的合成周期
[0051]
如图1所示的合成路线,在一干净250ml玻璃夹套反应瓶中加入80ml 0.1mol/l磷酸盐缓冲液,称取脂肪酶(0.5g)加入到磷酸盐缓冲液中搅拌均匀。将反应体系控温在30℃。滴加底物(10g)的正己烷(20ml)溶液,开始反应。反应过程中,利用0.5mol/l naoh溶液控制ph在8左右。20小时后终止反应,将反应ph调至9,将反应液用硅藻土过滤,滤饼用乙酸乙酯淋洗,滤液用乙酸乙酯萃取。然后将水相调至ph 3,用乙酸乙酯萃取,将调酸后所得有机相合并,加入无水硫酸钠除水,再过滤获得有机相,减压浓缩得到粗品(s)-2-(4-溴苯基)丙酸4.9g。然后将粗品用0.5倍体积甲叔醚打浆过滤,滤饼干燥得到纯品(s)-2-(4-溴苯基)丙酸4.76g。
[0052]
由实施例5和实施例6可以看到,ph的调整,可缩短产物的合成周期,加快反应速率,另外,本发明提供的制备方法,可根据ph变化情况适时补加一定量的酶,进一步起到降低总反应时长,缩短周期的作用。
[0053]
实施例7(r)-2-(4-溴苯基)丙酸甲酯的回收
[0054]
如图1所示的合成路线,在一干净250ml玻璃反应瓶中加入100ml甲醇,称取50g(r)-2-(4-溴苯基)丙酸甲酯,搅拌至溶解。向反应液中加入meona(100g),加热回流反应15小时后终止反应。过滤,将滤液旋干得到50g 2-(4-溴苯基)丙酸甲酯。
[0055]
综上所述,上述各实施例仅为本发明的较佳实施例而已,并不用以限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,皆应包含在本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1