一种室温下稳定的有机双自由基单分子器件及制备方法
1.本发明涉及有机双自由基单分子器件领域,特别地涉及一种室温下稳定的有机双自由基单分子器件及制备方法。
背景技术:
2.随着分子电子学的快速发展,单分子器件这一概念也被研究者提及,在此基础上通过实验与理论计算相结合,许多较为复杂的问题得以深入的研究,如单分子化学结构(几何长度、共轭程度等)对电荷输运的影响,分子与电极的连接界面问题,电子在分子结传输过程中的量子干涉效应(quantum interference,qi)等。有机自由基(organic radicals)是具有未配对电子的有机小分子或者聚合物。自由基中的未配对电子处于最外层分子轨道,极易发生电子的得失,因此有机自由基通常具有活泼的化学性质,如分子自身容易发生氧化、聚合等化学反应,导致大多数自由基的寿命很短。
技术实现要素:
3.针对上述现有技术中的问题,本技术提出了一种室温下稳定的有机双自由基单分子器件,包括锚定基团和双自由基,所述双自由基含有两个未成对电子。
4.优选地,有机双自由基单分子器件的分子式为:
[0005][0006]
优选地,两个未成对电子通过氧化反应获得。
[0007]
本技术还涉及一种室温下稳定的有机双自由基单分子器件的制备方法,包括以下步骤:
[0008]
步骤s1、取1,4-二溴苯、三二亚苄基丙酮二钯、三叔丁基四氟硼酸盐和叔丁醇钠,将所有反应物固体和搅拌用磁子混合后,密封置于冷凝回流系统中抽充氮气,加入第一溶剂,油浴加热并开启磁力搅拌,停止加热后,让整个反应体系缓慢冷却至室温后再处理;
[0009]
步骤s2、萃取反应液,取有机层溶液,将萃取得到的溶液除水、过滤后,去掉第一溶剂,得到第一固体粗产物,通过色谱柱提纯得到粗产物,淋洗剂为正己烷和/或二氯甲烷;
[0010]
步骤s3、将第一固体粗产物和搅拌用磁子混合,加入第二溶剂,开启磁力搅拌,反应后,过滤并去掉第二溶剂,得到第二固体粗产物;
[0011]
步骤s4、通过色谱柱提纯得到的第三粗产物,淋洗剂为二氯甲烷,得到黑色粉末。
[0012]
优选地,1,4-二溴苯的用量为“235mg,1mmol,1.0eq”、三二亚苄基丙酮二钯的用量为“45mg,0.05mmol,0.05eq”、三叔丁基四氟硼酸盐的用量为“28mg,0.1mmol,0.1eq”和叔丁醇钠的用量为“192mg,2mmol,2eq”。
[0013]
优选地,淋洗剂为正己烷:二氯甲烷=3:1、2:1或1:1。
[0014]
优选地,第一溶剂为347mg、2.5mmol、2.5eq的4-硫甲基苯胺和30毫升甲苯。
[0015]
优选地,第二溶剂为10毫升二氯甲烷。
[0016]
本技术还涉及一种室温下稳定的有机双自由基单分子器件的制备方法,包括以下步骤:
[0017]
步骤s1、取4,4-二溴联苯、三二亚苄基丙酮二钯、三叔丁基四氟硼酸盐和叔丁醇钠,将所有反应物固体和搅拌用磁子混合后,密封置于冷凝回流系统中抽充氮气,加入第一溶剂,油浴加热并开启磁力搅拌,停止加热后,让整个反应体系缓慢冷却至室温后再处理;
[0018]
步骤s2、萃取反应液,取有机层溶液,将萃取得到的溶液除水、过滤后,去掉第一溶剂,得到第一固体粗产物,通过色谱柱提纯得到粗产物,淋洗剂为正己烷和/或二氯甲烷;
[0019]
步骤s3、将第一固体粗产物和搅拌用磁子混合,加入第二溶剂,开启磁力搅拌,反应后,过滤并去掉第二溶剂,得到第二固体粗产物;
[0020]
步骤s4、通过色谱柱提纯得到的第三粗产物,淋洗剂为二氯甲烷,得到黑色粉末。
[0021]
本技术还涉及一种室温下稳定的有机双自由基单分子器件的制备方法,其特征在于,包括以下步骤:
[0022]
步骤s1、取2,7-二溴-9,9-二甲基芴、三二亚苄基丙酮二钯、三叔丁基四氟硼酸盐和叔丁醇钠,将所有反应物固体和搅拌用磁子混合后,密封置于冷凝回流系统中抽充氮气,加入第一溶剂,油浴加热并开启磁力搅拌,停止加热后,让整个反应体系缓慢冷却至室温后再处理;
[0023]
步骤s2、萃取反应液,取有机层溶液,将萃取得到的溶液除水、过滤后,去掉第一溶剂,得到第一固体粗产物,通过色谱柱提纯得到粗产物,淋洗剂为正己烷和/或二氯甲烷;
[0024]
步骤s3、将第一固体粗产物和搅拌用磁子混合,加入第二溶剂,开启磁力搅拌,反应后,过滤并去掉第二溶剂,得到第二固体粗产物;
[0025]
步骤s4、通过色谱柱提纯得到的第三粗产物,淋洗剂为二氯甲烷,得到黑色粉末。
[0026]
上述技术特征可以各种适合的方式组合或由等效的技术特征来替代,只要能够达到本发明的目的。
[0027]
本发明提供的一种室温下稳定的有机双自由基单分子器件及制备方法,与现有技术相比,至少具备有以下有益效果:有机共轭双自由基与单自由基相比,我们探究重点的不同之处在于对两个未成对电子在单分子输运的影响作用。本发明构筑的有机双自由基单分子器件能在室温条件下稳定,且化合物氧化前后的电导值变化明显,这在一定程度上证明未配对电子的离域范围能提升单分子电导。
附图说明
[0028]
在下文中将基于实施例并参考附图来对本发明进行更详细的描述。其中:
[0029]
图1显示了ph-nh-o合成路线图;
[0030]
图2显示了2ph-nh-o合成路线图;
[0031]
图3显示了wu-nh-o合成路线图;
[0032]
图4显示了me-nh-o合成路线图;
[0033]
图5显示了3ph-nh-o合成路线图;
[0034]
图6显示了stm-bj连接示意图;
[0035]
图7显示了ph-nh和ph-nh-o溶液的紫外-可见光-近红外吸收光谱图;
[0036]
图8显示了ph-nh和ph-nh-o的一维电导直方图;
[0037]
图9显示了2ph-nh-o的电子自旋共振谱图;
[0038]
图10显示了2ph-nh和2ph-nh-o溶液的紫外-可见光-近红外吸收光谱图;
[0039]
图11显示了2ph-nh和2ph-nh-o的一维电导直方图;
[0040]
图12显示了wu-nh和wu-nh-o溶液的紫外-可见光-近红外吸收光谱图;
[0041]
图13显示了wu-nh和wu-nh-o的一维电导直方图;
具体实施方式
[0042]
下面将结合附图对本发明作进一步说明。
[0043]
本发明提供了一种室温下稳定的有机双自由基单分子器件及制备方法。
[0044]
实施例一
[0045]
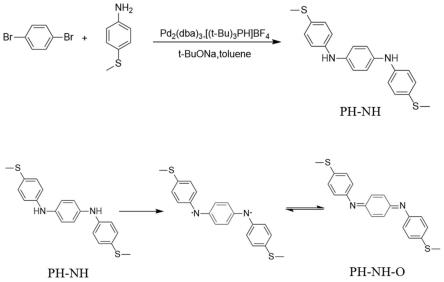
如图1所示,首先,制备ph-nh的具体步骤如下:取1,4-二溴苯(235mg,1mmol,1.0eq)、三(二亚苄基丙酮二钯)(45mg,0.05mmol,0.05eq)、三叔丁基四氟硼酸盐(28mg,0.1mmol,0.1eq)和叔丁醇钠(192mg,2mmol,2eq)。将所有反应物固体和搅拌用磁子依次加入100毫升的双口茄形瓶后,将其密封置于冷凝回流系统中抽充氮气三次,使用注射器加入4-硫甲基苯胺(347mg,2.5mmol,2.5eq)和30毫升甲苯作为溶剂,油浴加热至110摄氏度并开启磁力搅拌,反应24个小时后停止加热,让整个反应体系缓慢冷却至室温后再处理。使用乙酸乙酯与饱和食盐水萃取反应液,取上层有机层溶液,并重复萃取操作三次。萃取得到的溶液使用足量的无水硫酸钠除水并过滤后,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过硅胶色谱柱提纯得到的粗产物,淋洗剂为正己烷:二氯甲烷=3:1,得到ph-nh。
[0046]
ph-nh-o的合成及当量比:反应物及当量比为:产物ph-nh(100mg,0.28mmol,1.0eq),二氧化铅(680mg,2.8mmol,10.0eq)。将所有反应物固体和搅拌用磁子依次加入50毫升的单口茄形瓶后,加入10毫升二氯甲烷作为溶剂,开启磁力搅拌,反应半个小时后,在滤纸上平铺适量硅藻土并将反应液过滤,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过中性三氧化二铝色谱柱提纯得到的粗产物,淋洗剂为二氯甲烷。得到83mg黑色粉末,即为ph-nh-o,产率为83%。
[0047]
实施例二
[0048]
如图2所示,首先,制备ph-nh的具体步骤如下:取4,4-二溴联苯(235mg,1mmol,1.0eq)、三(二亚苄基丙酮二钯)(45mg,0.05mmol,0.05eq)、三叔丁基四氟硼酸盐(28mg,0.1mmol,0.1eq)和叔丁醇钠(192mg,2mmol,2eq)。将所有反应物固体和搅拌用磁子依次加入100毫升的双口茄形瓶后,将其密封置于冷凝回流系统中抽充氮气三次,使用注射器加入4-硫甲基苯胺(347mg,2.5mmol,2.5eq)和30毫升甲苯作为溶剂,油浴加热至110摄氏度并开启磁力搅拌,反应24个小时后停止加热,让整个反应体系缓慢冷却至室温后再处理。使用乙
酸乙酯与饱和食盐水萃取反应液,取上层有机层溶液,并重复萃取操作三次。萃取得到的溶液使用足量的无水硫酸钠除水并过滤后,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过硅胶色谱柱提纯得到的粗产物,淋洗剂为正己烷:二氯甲烷=2:1,得到2ph-nh。
[0049]
2ph-nh-o的合成及当量比:反应物及当量比为:产物2ph-nh(100mg,0.23mmol,1.0eq),二氧化铅(547mg,2.3mmol,10.0eq)。将所有反应物固体和搅拌用磁子依次加入50毫升的单口茄形瓶后,加入10毫升二氯甲烷作为溶剂,开启磁力搅拌,反应半个小时后,在滤纸上平铺适量硅藻土并将反应液过滤,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过中性三氧化二铝色谱柱提纯得到的粗产物,淋洗剂为二氯甲烷。得到30mg黑色粉末,即为2ph-nh-o,产率为30%。
[0050]
实施例三
[0051]
如图3所示,首先,制备wu-nh的具体步骤如下:取2,7-二溴-9,9-二甲基芴(235mg,1mmol,1.0eq)、三(二亚苄基丙酮二钯)(45mg,0.05mmol,0.05eq)、三叔丁基四氟硼酸盐(28mg,0.1mmol,0.1eq)和叔丁醇钠(192mg,2mmol,2eq)。将所有反应物固体和搅拌用磁子依次加入100毫升的双口茄形瓶后,将其密封置于冷凝回流系统中抽充氮气三次,使用注射器加入4-硫甲基苯胺(347mg,2.5mmol,2.5eq)和30毫升甲苯作为溶剂,油浴加热至110摄氏度并开启磁力搅拌,反应24个小时后停止加热,让整个反应体系缓慢冷却至室温后再处理。使用乙酸乙酯与饱和食盐水萃取反应液,取上层有机层溶液,并重复萃取操作三次。萃取得到的溶液使用足量的无水硫酸钠除水并过滤后,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过硅胶色谱柱提纯得到的粗产物,淋洗剂为正己烷:二氯甲烷=1:1,得到wu-nh。
[0052]
wu-nh-o的合成及当量比:反应物及当量比为:产物wu-nh(100mg,0.21mmol,1.0eq),二氧化铅(501mg,2.1mmol,10.0eq)。将所有反应物固体和搅拌用磁子依次加入50毫升的单口茄形瓶后,加入10毫升二氯甲烷作为溶剂,开启磁力搅拌,反应半个小时后,在滤纸上平铺适量硅藻土并将反应液过滤,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过中性三氧化二铝色谱柱提纯得到的粗产物,淋洗剂为二氯甲烷。得到28mg黑色粉末,即为wu-nh-o,产率为28%。
[0053]
实施例四
[0054]
如图4所示,首先,制备me-nh的具体步骤如下:取1,4-二溴苯(235mg,1mmol,1.0eq)、三(二亚苄基丙酮二钯)(45mg,0.05mmol,0.05eq)、三叔丁基四氟硼酸盐(28mg,0.1mmol,0.1eq)和叔丁醇钠(192mg,2mmol,2eq)。将所有反应物固体和搅拌用磁子依次加入100毫升的双口茄形瓶后,将其密封置于冷凝回流系统中抽充氮气三次,使用注射器加入4-硫甲基苯胺(347mg,2.5mmol,2.5eq)和30毫升甲苯作为溶剂,油浴加热至110摄氏度并开启磁力搅拌,反应24个小时后停止加热,让整个反应体系缓慢冷却至室温后再处理。使用乙酸乙酯与饱和食盐水萃取反应液,取上层有机层溶液,并重复萃取操作三次。萃取得到的溶液使用足量的无水硫酸钠除水并过滤后,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过硅胶色谱柱提纯得到的粗产物,淋洗剂为正己烷:二氯甲烷=2:1,得到me-nh。
[0055]
me-nh-o的合成及当量比:反应物及当量比为:产物me-nh(100mg,0.28mmol,1.0eq),二氧化铅(680mg,2.8mmol,10.0eq)。将所有反应物固体和搅拌用磁子依次加入50毫升的单口茄形瓶后,加入10毫升二氯甲烷作为溶剂,开启磁力搅拌,反应半个小时后,在滤纸上平铺适量硅藻土并将反应液过滤,用旋转蒸发仪在低压下去掉溶剂得到固体粗产
物。通过中性三氧化二铝色谱柱提纯得到的粗产物,淋洗剂为二氯甲烷。得到20mg黑色粉末,即为me-nh-o,产率为20%。
[0056]
实施例五
[0057]
如图5所示,首先,制备ph-nh的具体步骤如下:取4,4-二溴三联苯(235mg,1mmol,1.0eq)、三(二亚苄基丙酮二钯)(45mg,0.05mmol,0.05eq)、三叔丁基四氟硼酸盐(28mg,0.1mmol,0.1eq)和叔丁醇钠(192mg,2mmol,2eq)。将所有反应物固体和搅拌用磁子依次加入100毫升的双口茄形瓶后,将其密封置于冷凝回流系统中抽充氮气三次,使用注射器加入4-硫甲基苯胺(347mg,2.5mmol,2.5eq)和30毫升甲苯作为溶剂,油浴加热至110摄氏度并开启磁力搅拌,反应24个小时后停止加热,让整个反应体系缓慢冷却至室温后再处理。使用乙酸乙酯与饱和食盐水萃取反应液,取上层有机层溶液,并重复萃取操作三次。萃取得到的溶液使用足量的无水硫酸钠除水并过滤后,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过硅胶色谱柱提纯得到的粗产物,淋洗剂为正己烷:二氯甲烷=1:1,得到3ph-nh。
[0058]
3ph-nh-o的合成及当量比:反应物及当量比为:产物3ph-nh(100mg,0.28mmol,1.0eq),二氧化铅(680mg,2.8mmol,10.0eq)。将所有反应物固体和搅拌用磁子依次加入50毫升的单口茄形瓶后,加入10毫升二氯甲烷作为溶剂,开启磁力搅拌,反应半个小时后,在滤纸上平铺适量硅藻土并将反应液过滤,用旋转蒸发仪在低压下去掉溶剂得到固体粗产物。通过中性三氧化二铝色谱柱提纯得到的粗产物,淋洗剂为二氯甲烷。得到54mg黑色粉末,即为3ph-nh-o,产率为54%。
[0059]
如图6所示,将装有金电极的stm-bj探针与附着于金基底的分子相接触,在外加电场的作用下,stm-bj探针被均匀缓慢提升,分子也逐渐被拉起来。在这个过程中可以看到电导变化的2个阶段:(1)随着金电极的提升,电导呈台阶式下降,通过一维电导直方图可以看到电导峰;(2)形成金电极-分子-金基底的结构,并由此得出分子的电导值。stmbj可以通过反复移动针尖使其与吸附有样品分子的基底电极接触和分离,来快速创建数千个分子结。当针尖距离基底足够近时,分子有机会将针尖电极和基底电极连接起来。然后向上提拉针尖,以断开金属-分子-金属连接。通过精确控制针尖的移动,即改变针尖与基底之间的距离,可以改变桥接分子的数量。
[0060]
通过改变苯基苯胺结构中心结构的苯环数量或者增加甲基的方式来改变电子传输分布,能够增加自由基浓度。
[0061]
ph-nh-o的理论计算结果为y=0,初步说明ph-nh-o应该为醌式结构,这一点通过esr可佐证,因为ph-nh-o的esr信号很弱,几乎可以忽略不计。除此之外,我们对该分子进行了吸收光谱的测试,如图7所示。从图7中我们可以看到ph-nh-o在463nm附近只有一个最大吸收峰。
[0062]
为了探究有机双自由基单分子电荷输运机理,我们对ph-nh和ph-nh-o进行了stm-bj测试,结果如图8所示。
[0063]
我们可以发现ph-nh-o分子出现了两个电导峰。与ph-nh的电导值相比,表现为一个增大、一个减小的趋势,推测是由于ph-nh-o出现了同分异构体。这一点我们通过核磁共振氢谱证明了ph-nh-o确实是有同分异构体的存在。
[0064]
2ph-nh-o的理论计算结果为y=0.473,初步说明2ph-nh-o应该为自由基结构,这一点通过esr可佐证,图中可以清楚看见2ph-nh-o出现超精细裂分的情况,初步说明该分子
中具有两个未成对电子,为双自由基结构,如图9所示。除此之外,我们对该分子进行了吸收光谱的测试,如图10所示。从图10中我们可以看到2ph-nh-o溶液的最大吸收峰处于438nm的紫外波段,并在590nm的可见光波段有一个相对较低的吸收峰,未配对电子通常会带来在可见光甚至是近红外波段的吸收,因此从2ph-nh-o溶液的吸收光谱可以初步推测具有未配对电子。
[0065]
为了探究有机双自由基单分子电荷输运机理,我们对2ph-nh和2ph-nh-o进行了stm-bj测试,结果如图11所示。
[0066]
我们可以发现氧化后的电导值相较于氧化前有所提高,初步推测是因为单分子结中未配对电子所带来的影响。
[0067]
wu-nh-o的理论计算结果为y=0.03,初步推测wu-nh-o应该为自由基结构,之后会进行esr测试进一步确认。除此之外,我们对该分子进行了吸收光谱的测试,如图12所示。从图12中我们可以看到wu-nh-o溶液的最大吸收峰处于361nm的紫外波段,并在759nm的可见光波段有一个相对较低的吸收峰,未配对电子通常会带来在可见光甚至是近红外波段的吸收,因此从wu-nh-o溶液的吸收光谱可以初步推测具有未配对电子。
[0068]
为了探究有机双自由基单分子电荷输运机理,我们对wu-nh和wu-nh-o进行了stm-bj测试,结果如图13所示。
[0069]
我们可以发现氧化后的电导值相较于氧化前有所提高,初步推测是因为单分子结中未配对电子所带来的影响。除此之外,2ph-nh-o和wu-nh-o的电导值差不多,但是氧化之前2ph-nh和wu-nh的电导相差较大。初步推测分子位阻会减小电导,而在氧化之后由于电子离域范围的增加,使得分子位阻对电导的影响可以忽略不计。
[0070]
虽然在本文中参照了特定的实施方式来描述本发明,但是应该理解的是,这些实施例仅仅是本发明的原理和应用的示例。因此应该理解的是,可以对示例性的实施例进行许多修改,并且可以设计出其他的布置,只要不偏离所附权利要求所限定的本发明的精神和范围。应该理解的是,可以通过不同于原始权利要求所描述的方式来结合不同的从属权利要求和本文中所述的特征。还可以理解的是,结合单独实施例所描述的特征可以使用在其他所述实施例中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1