一种用于痕量检测与吸附Fe的制作方法
一种用于痕量检测与吸附fe
3+
离子的双功能材料及制备方法
技术领域
1.本发明属于重金属离子吸附与检测领域,具体涉及一种基于碳点修饰的双功能材料,并将其应用于fe
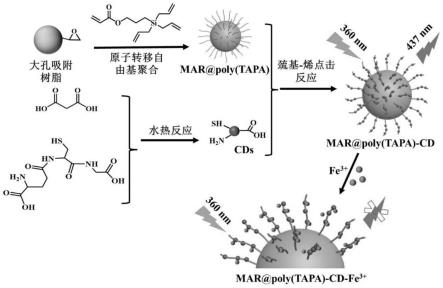
3+
离子的痕量检测与吸附。
背景技术:
2.铁元素广泛存在于自然环境中,是人体正常细胞和机体功能所必需的元素。然而,人体内铁的浓度不合理与各种疾病有关。体内铁水平低可导致缺铁性贫血,但异常高铁水平与各种疾病有关,如器官损伤、关节炎、肝病、心脏异常等。此外,饮用水中fe
3+
离子的大量积累会导致变色和异味,进而造成水污染(文献1,c.li,et al"chitosan based macromolecular probes for the selectivedetection and removal of fe
3+
ion"int.j.biol.macromol.2021,186,303-313)。因此,研究水环境中fe
3+
离子的有效检测和吸附策略具有重要意义。
3.传统的fe
3+
离子检测方法包括原子吸收光谱法、分子吸收分光光度法、电感耦合等离子质谱法等,这些技术通常用于金属离子的测定,但其需要昂贵的仪器,熟练的操作技能,复杂和耗时的操作过程(文献2,s.chakraborty,et al "detection of iron(iii)by chemo and fluoro-sensing technology"inorg.chem. commun.2020,121,108189)。
4.碳点(cd)具有生物相容性好、表面官能团丰富、毒性低等优点,是一种很有前途的荧光纳米材料。cds的表征尺寸小于10nm,具有良好的光致发光性能。自2004年提出cds概念以来,它被广泛应用于生物传感、催化、生物医学、生物成像等领域(文献3,c.hu,et al"a simple one-step synthesis ofmelanin-originated red shift emissive carbonaceous dots forbioimaging"j.colloid. interf.sci.2016,480,85-90)。此外,cds具有较高的灵敏度和选择性,在金属离子检测领域得到了广泛的应用。
5.迄今为止,大孔吸附树脂(mar)因其优异的机械强度、高孔隙率、稳定的化学性质和可到达的吸附位点,在分离和吸附领域得到了广泛的应用(文献4, j.li,et al"development of adsorptive(non-ionic)macroporous resins and theiruses in the purification of pharmacologically-active natural products from plantsources"nat.prod.rep.2010,27,1493-1
510)。
技术实现要素:
6.本发明合成了大孔吸附树脂作为载体,并通过表面引发原子转移自由基聚合(si-atrp)技术将含有三个乙烯基和一个丙烯酸基团的3-(三烯丙基硅基)丙基丙烯酸酯(tapa)接枝到大孔吸附树脂表面。由于含有大量乙烯基,蓝色荧光cds可以通过巯基-烯反应被固定在mar@poly(tapa)表面,之后将这种双功能材料命名为mar@poly(tapa)-cd,并且可以应用于fe
3+
离子的荧光检测和吸附。本发明功能材料的开发对去除水环境中的重金属离子具有重要意义。
7.本发明的一种用于痕量检测与吸附fe
3+
离子的双功能材料的制备方法,包括以下
3-基甲基丙烯酸酯、乙二醇二甲基丙烯酸酯、甲苯与环己醇的混合比例为 0.3~0.5g/6~8ml/6~8ml/6~8ml/6~8ml。
22.本发明优选地,在步骤s2中,二氯甲烷溶液与三乙胺的混合比例为30~ 60ml/1~2ml,且添加三乙胺后继续搅拌10~20min;所述三乙胺、2-溴异丁酰溴与4-二甲基氨基吡啶的混合比例为1~2ml/0.5~1ml/10~20mg。
23.本发明还提供了一种用于痕量检测与吸附fe
3+
离子的双功能材料,结构为:带有环氧的大孔吸附树脂微球表面接枝含有三个乙烯基双键和一个丙烯酸双键的功能单体3-(三烯丙基硅烷基)丙烯酸丙酯(tapa)的聚合物刷,从而形成带有聚合物刷的材料mar@poly(tapa),并在所述聚合物刷上接枝带有巯基的碳点cd材料得到用于吸附和检测的双功能材料 mar@poly(tapa)-cd;
24.所述带有环氧的大孔吸附树脂微球为以7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯为功能单体,乙二醇二甲基丙烯酸酯为交联剂,利用种子溶胀法制备得到的带有环氧官能团的大孔吸附树脂。
[0025]
本发明中将含有三个乙烯基双键和一个丙烯酸双键的功能单体3-(三烯丙基硅烷基)丙烯酸丙酯(tapa)的聚合物刷接枝在带有环氧的大孔吸附树脂微球表面的方法是表面引发原子转移自由基聚合(si-atrp)技术。
[0026]
本发明中所述碳点cd为以丙二酸和谷胱甘肽为前驱体制备的一种具有蓝色发射的碳点cd。
[0027]
本发明还提供了一种所述的用于痕量检测与吸附fe
3+
离子的双功能材料的应用,应用于水环境中fe
3+
离子的检测与吸附。
[0028]
本发明的双功能材料mar@poly(tapa)-cd以7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯和乙二醇二甲基丙烯酸酯分别作为功能单体和交联剂制备带有环氧基团的大孔吸附树脂为底物。
[0029]
然后,通过表面引发原子转移自由基聚合(si-atrp)将含有三个烯基的功能单体3-(三烯丙基硅基)丙烯酸丙酯接枝大孔吸附树脂到表面。
[0030]
随后,以丙二酸和谷胱甘肽为前驱体制备了一种具有蓝色发射的碳点 (cd),并将其固定在大孔吸附树脂表面形成双功能树脂材料,命名为 mar@poly(tapa)-cd。
[0031]
由于mar@poly(tapa)-cd具有荧光特性,对金属离子特别是fe
3+
离子具有不同程度的荧光猝灭,猝灭程度达到46.2%。在0~80nmol l-1
的线性范围内(相关系数为0.9909),检出限为14.51nmol l-1
,可用于痕量检测。此外,功能化mar@poly(tapa)-cd作为吸附和分离的优良载体,也用于吸附fe
3+
离子,具有32.9mg g-1
的高吸附量。吸附fe
3+
的平衡时间为45min。因此,该双功能材料可以同时实现水溶液中fe
3+
离子的检测和吸附,具有很大的商业化潜力。
[0032]
本发明制备的双功能检测与吸附材料,对fe
3+
离子具有痕量检测的能力,并且对水质中的fe
3+
离子具备较好的吸附能力。本发明基于碳点悬浮液,提供了一种利用荧光检测测定吸附量的策略,相比于其他检测手段,具有操作方便,花费较少等优点。另外,对于本发明的制备方法:其原料易得、成本低廉、步骤简单、反应条件温和,能有效适合于大规模制备。
附图说明
[0033]
图1为本发明制备的双功能材料的制备流程图以及荧光猝灭机理图;
[0034]
图2为本发明制备的大孔吸附树脂和双功能材料mar@poly(tapa)-cd 的sem图像;
[0035]
图3为本发明初始的大孔吸附树脂和双功能材料mar@poly(tapa)-cd 的氮气吸附/脱附等温曲线(a),孔径分布图(b);
[0036]
图4为不同金属离子对双功能材料mar@poly(tapa)-cd的(a)荧光猝灭谱图(b)淬灭程度以及测定fe
3+
离子的(c)荧光淬灭谱图和(d)定量测量曲线;
[0037]
图5为双功能材料mar@poly(tapa)-cd对fe
3+
离子的等温吸附曲线。
具体实施方式
[0038]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0039]
实施例1
[0040]
以双功能材料(mar@poly(tapa)-cd)为基质,用于水质中fe
3+
离子的检测与吸附。
[0041]
s0碳点材料的制备
[0042]
称取0.5g的谷胱甘肽,0.34g的丙二酸,将二者充分溶解在25ml的去离子水中,在180℃下一步水热反应2h,过滤后得到含有巯基,氨基和羧基官能团的碳点悬浮液。
[0043]
s1.制备单分散的大孔吸附树脂(mar)微球
[0044]
首先将10ml的7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯单体加入到2%的聚乙烯吡咯烷酮溶液中(2g聚乙烯吡咯烷酮(pvp)溶解于87.8ml乙醇溶液中),之后添加0.2g偶氮二异丁腈(aibn),通氮除氧30min后在70℃的温度及80r/min的转速下旋蒸加热反应24h;反应结束后,利用无水乙醇离心洗涤反应后的产物,然后置于浓度为0.2%的十二烷基硫酸钠(sds)溶液中,4℃环境下静置得到种子溶液;
[0045]
取0.36g偶氮二异丁腈(aibn)置于烧杯中,然后加入6ml 7-氧杂双环 [4.1.0]庚-3-基甲基丙烯酸酯、6ml乙二醇二甲基丙烯酸酯、6ml甲苯与6ml 环己醇混合组成油相;
[0046]
以浓度为5%的聚乙烯醇(pva)和浓度为0.2%的十二烷基硫酸钠(sds) 的混合液为水相;
[0047]
将油相与水相混合并置于细胞破碎仪中充分乳化,得到乳液;
[0048]
取8.2ml的种子溶液置于烧瓶中,室温条件下以200r/min的转速磁力搅拌30min后加入乳液,接着通氮除氧30min并继续磁力搅拌24h(以此使油相被充分吸收到种子内部),然后升温至70℃并保温反应24h,反应后其聚合反应产物经48h索提、无水乙醇和丙酮依次洗涤、60℃真空干燥12h得到单分散的大孔吸附树脂(mar)微球;
[0049]
s2.微球表面接枝聚合物刷形成聚合基质
[0050]
将2g大孔吸附树脂(mar)微球分散于36ml浓度为0.1mol/l硫酸溶液中,60℃条件下反应12h,然后将反应后的产物洗涤至中性并在60℃下真空干燥12h,得到酸化的mar;
[0051]
将1g酸化的mar加入至32ml的二氯甲烷溶液中,200r/min的速度下冰浴搅拌30min,然后加入1ml三乙胺并继续搅拌10min,接着在依次加入0.8ml 的2-溴异丁酰溴和
15.2mg的4-二甲基氨基吡啶,维持冰浴反应2h后转移至室温条件下反应24h,将反应后的产物依次用二氯甲烷和水各洗涤3次,之后在60℃下真空干燥12h,得到大分子引发剂;atrp反应:将1g大分子引发剂、25.8mg的2,2'-联吡啶和1ml 3-(三烯丙基硅基)丙烯酸丙酯(tapa)依次分散于10ml乙醇-水的混合溶剂中,其中乙醇-水的混合溶剂中乙醇/水的体积比为4/1;
[0052]
循环执行3次冷冻、抽真空和充氮的操作以去除溶剂中的氧气,然后添加 26mg的溴化亚铜催化剂;
[0053]
再循环执行3次冷冻、抽真空和充氮的操作并且在氮气氛围下60℃催化反应24h,得到表面接枝有聚合物刷的聚合基质(mar@poly(tapa)微球);
[0054]
s3.所述聚合基质表面碳点的接枝
[0055]
取10ml含有巯基的碳点悬浮液,加入盛有300mg mar@poly(tapa) 的烧杯中,保持250rpm的转速,常温下在365nm紫外灯的照射下反应2h。之后将产物用乙醇和去离子水分别洗涤3次,40℃干燥12h后得到接枝有碳点的双功能大孔吸附树脂材料。其为mar@poly(tapa)-cd微球材料,且微球的粒径为5~10μm,平均粒径为6μm。
[0056]
实施例2
[0057]
以双功能材料(mar@poly(tapa)-cd)为基质,用于水质中fe
3+
离子的检测与吸附。
[0058]
1、碳点材料的制备
[0059]
称取0.85g的谷胱甘肽,0.30g的丙二酸,将二者充分溶解在20ml的去离子水中,在160℃下一步水热反应2.5h,过滤后得到含有巯基,氨基和羧基官能团的碳点悬浮液。
[0060]
2、双功能材料(mar@poly(tapa)-cd)的制备
[0061]
s1.制备单分散的大孔吸附树脂(mar)微球
[0062]
首先将5ml的7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯单体加入到2%的聚乙烯吡咯烷酮溶液中(2g聚乙烯吡咯烷酮(pvp)溶解于87.8ml乙醇溶液中),之后添加0.1g偶氮二异丁腈(aibn),通氮除氧30min后在70℃的温度及 80r/min的转速下旋蒸加热反应20h;反应结束后,利用无水乙醇离心洗涤反应后的产物,然后置于浓度为0.2%的十二烷基硫酸钠(sds)溶液中,4℃环境下静置得到种子溶液;
[0063]
取0.36g偶氮二异丁腈(aibn)置于烧杯中,然后加入8ml 7-氧杂双环 [4.1.0]庚-3-基甲基丙烯酸酯、8ml乙二醇二甲基丙烯酸酯、8ml甲苯与8ml 环己醇混合组成油相;
[0064]
以浓度为2%的聚乙烯醇(pva)和浓度为0.25%的十二烷基硫酸钠(sds) 的混合液为水相;
[0065]
将油相与水相混合并置于细胞破碎仪中充分乳化,得到乳液;
[0066]
取8.0ml的种子溶液置于烧瓶中,室温条件下以200r/min的转速磁力搅拌30min后加入乳液,接着通氮除氧30min并继续磁力搅拌24h(以此使油相被充分吸收到种子内部),然后升温至70℃并保温反应24h,反应后其聚合反应产物经40h索提、无水乙醇和丙酮依次洗涤、70℃真空干燥20h得到单分散的大孔吸附树脂(mar)微球;
[0067]
s2.微球表面接枝聚合物刷形成聚合基质
[0068]
将1g大孔吸附树脂(mar)微球分散于20ml浓度为0.1mol/l硫酸溶液中,40℃条件下反应12h,然后将反应后的产物洗涤至中性并在60℃下真空干燥12h,得到酸化的mar;
[0069]
将1g酸化的mar加入至30ml的二氯甲烷溶液中,200r/min的速度下冰浴搅拌
30min,然后加入1.5ml三乙胺并继续搅拌10min,接着在依次加入1ml 的2-溴异丁酰溴和15mg的4-二甲基氨基吡啶,维持冰浴反应2h后转移至室温条件下反应24h,将反应后的产物依次用足量的二氯甲烷和水各洗涤3次,之后在60℃下真空干燥12h,得到大分子引发剂;
[0070]
atrp反应:将0.5g大分子引发剂、20mg的2,2'-联吡啶和.05ml 3-(三烯丙基硅基)丙烯酸丙酯(tapa)依次分散于10ml乙醇-水的混合溶剂中,其中乙醇-水的混合溶剂中乙醇/水的体积比为4/1;
[0071]
循环执行3次冷冻、抽真空和充氮的操作以去除溶剂中的氧气,然后添加 20mg的溴化亚铜催化剂/10ml溶剂;
[0072]
再循环执行3次冷冻、抽真空和充氮的操作并且在氮气氛围下60℃催化反应24h,得到表面接枝有聚合物刷的聚合基质(mar@poly(tapa)微球);
[0073]
s3.所述聚合基质表面碳点的接枝
[0074]
取5ml含有巯基的碳点悬浮液,加入盛有100mg mar@poly(tapa)的烧杯中,保持250rpm的转速,常温下在365nm紫外灯的照射下反应2h。之后将产物用乙醇和去离子水分别洗涤3次,40℃干燥12h后得到接枝有碳点的双功能大孔吸附树脂材料。其为mar@poly(tapa)-cd微球材料,且微球的粒径为5~10μm,平均粒径为6μm。
[0075]
实施例3
[0076]
以双功能材料(mar@poly(tapa)-cd)为基质,用于水质中fe
3+
离子的检测与吸附。
[0077]
s0.碳点材料的制备
[0078]
称取1g的谷胱甘肽,0.6g的丙二酸,将二者充分溶解在30ml的去离子水中,在200℃下一步水热反应4h,过滤后得到含有巯基,氨基和羧基官能团的碳点悬浮液。
[0079]
s1.制备单分散的大孔吸附树脂(mar)微球
[0080]
首先将15ml的7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯单体加入到5%的聚乙烯吡咯烷酮溶液中(5g聚乙烯吡咯烷酮(pvp)溶解于87.8ml乙醇溶液中),之后添加0.5g偶氮二异丁腈(aibn),通氮除氧30min后在80℃的温度及80r/min的转速下旋蒸加热反应20h;反应结束后,利用无水乙醇离心洗涤反应后的产物,然后置于浓度为0.5%的十二烷基硫酸钠(sds)溶液中,5℃环境下静置得到种子溶液;
[0081]
取0.5g偶氮二异丁腈(aibn)置于烧杯中,然后加入10ml 7-氧杂双环 [4.1.0]庚-3-基甲基丙烯酸酯、10ml乙二醇二甲基丙烯酸酯、10ml甲苯与10ml 环己醇混合组成油相;
[0082]
以浓度为5%的聚乙烯醇(pva)和浓度为0.5%的十二烷基硫酸钠(sds) 的混合液为水相;
[0083]
将油相与水相混合并置于细胞破碎仪中充分乳化,得到乳液;
[0084]
取12ml的种子溶液置于烧瓶中,室温条件下以250r/min的转速磁力搅拌60min后加入140ml乳液,接着通氮除氧并继续磁力搅拌24h(以此使油相被充分吸收到种子内部),然后升温至80℃并保温反应20h,反应后其聚合反应产物经48h索提、无水乙醇和丙酮依次洗涤、60℃真空干燥12h得到单分散的大孔吸附树脂(mar)微球;
[0085]
s2.微球表面接枝聚合物刷形成聚合基质
[0086]
将2g大孔吸附树脂(mar)微球分散于40ml浓度为0.3mol/l硫酸溶液中,60℃条件下反应12h,然后将反应后的产物洗涤至中性并在60℃下真空干燥12h,得到酸化的mar;
[0087]
将2g酸化的mar加入至60ml的二氯甲烷溶液中,300r/min的速度下冰浴搅拌
30min,然后加入3ml三乙胺并继续搅拌10min,接着在依次加入1.5ml 的2-溴异丁酰溴和20mg的4-二甲基氨基吡啶,维持冰浴反应3h后转移至室温条件下反应24h,将反应后的产物依次用二氯甲烷和水各洗涤3次,之后在 60℃下真空干燥12h,得到大分子引发剂;
[0088]
atrp反应:将2g大分子引发剂、30mg的2,2'-联吡啶和1ml 3-(三烯丙基硅基)丙烯酸丙酯(tapa)依次分散于10ml乙醇-水的混合溶剂中,其中乙醇-水的混合溶剂中乙醇/水的体积比为4/1;
[0089]
循环执行3次冷冻、抽真空和充氮的操作以去除溶剂中的氧气,然后添加 26mg的溴化亚铜催化剂/10ml溶剂;
[0090]
再循环执行3次冷冻、抽真空和充氮的操作并且在氮气氛围下50℃催化反应20h,得到表面接枝有聚合物刷的聚合基质(mar@poly(tapa)微球);
[0091]
s3.所述聚合基质表面碳点的接枝
[0092]
取10ml含有巯基的碳点悬浮液,加入盛有300mg mar@poly(tapa) 的烧杯中,保持300rpm的转速,常温下在365nm紫外灯的照射下反应2h。之后将产物用乙醇和去离子水分别洗涤3次,40℃干燥12h后得到接枝有碳点的双功能大孔吸附树脂材料。其为mar@poly(tapa)-cd微球材料,且微球的粒径为5~10μm,平均粒径为7μm。
[0093]
实施例4
[0094]
以双功能材料(mar@poly(tapa)-cd)为基质,用于水质中fe
3+
离子的检测与吸附。
[0095]
s0碳点材料的制备
[0096]
称取0.5g的谷胱甘肽,0.34g的丙二酸,将二者充分溶解在25ml的去离子水中,在180℃下一步水热反应2h,过滤后得到含有巯基,氨基和羧基官能团的碳点悬浮液。
[0097]
s1.制备单分散的大孔吸附树脂(mar)微球
[0098]
首先将10ml的7-氧杂双环[4.1.0]庚-3-基甲基丙烯酸酯单体加入到2%的聚乙烯吡咯烷酮溶液中(2g聚乙烯吡咯烷酮(pvp)溶解于87.8ml乙醇溶液中),之后添加0.2g偶氮二异丁腈(aibn),通氮除氧30min后在70℃的温度及80r/min的转速下旋蒸加热反应24h;反应结束后,利用无水乙醇离心洗涤反应后的产物,然后置于浓度为0.2%的十二烷基硫酸钠(sds)溶液中,4℃环境下静置得到种子溶液;
[0099]
取0.36g偶氮二异丁腈(aibn)置于烧杯中,然后加入8ml 7-氧杂双环 [4.1.0]庚-3-基甲基丙烯酸酯、8ml乙二醇二甲基丙烯酸酯、8ml甲苯与8ml 环己醇混合组成油相;
[0100]
以浓度为5%的聚乙烯醇(pva)和浓度为0.2%的十二烷基硫酸钠(sds) 的混合液为水相;
[0101]
将油相与水相混合并置于细胞破碎仪中充分乳化,得到乳液;
[0102]
取8.2ml的种子溶液置于烧瓶中,室温条件下以200r/min的转速磁力搅拌30min后加入乳液,接着通氮除氧30min并继续磁力搅拌24h(以此使油相被充分吸收到种子内部),然后升温至70℃并保温反应24h,反应后其聚合反应产物经48h索提、无水乙醇和丙酮依次洗涤、60℃真空干燥12h得到单分散的大孔吸附树脂(mar)微球;
[0103]
s2.微球表面接枝聚合物刷形成聚合基质
[0104]
将2g大孔吸附树脂(mar)微球分散于36ml浓度为0.1mol/l硫酸溶液中,60℃条件下反应12h,然后将反应后的产物洗涤至中性并在60℃下真空干燥12h,得到酸化的mar;
[0105]
将1g酸化的mar加入至32ml的二氯甲烷溶液中,200r/min的速度下冰浴搅拌
30min,然后加入2ml三乙胺并继续搅拌15min,接着在依次加入0.8ml 的2-溴异丁酰溴和20mg的4-二甲基氨基吡啶,维持冰浴反应2h后转移至室温条件下反应24h,将反应后的产物依次用二氯甲烷和水各洗涤3次,之后在60℃下真空干燥12h,得到大分子引发剂;atrp反应:将1g大分子引发剂、 25.8mg的2,2'-联吡啶和1ml 3-(三烯丙基硅基)丙烯酸丙酯(tapa)依次分散于 10ml乙醇-水的混合溶剂中,其中乙醇-水的混合溶剂中乙醇/水的体积比为 4/1;
[0106]
循环执行3次冷冻、抽真空和充氮的操作以去除溶剂中的氧气,然后添加 26mg的溴化亚铜催化剂;
[0107]
再循环执行3次冷冻、抽真空和充氮的操作并且在氮气氛围下60℃催化反应24h,得到表面接枝有聚合物刷的聚合基质(mar@poly(tapa)微球);
[0108]
s3.所述聚合基质表面碳点的接枝取10ml含有巯基的碳点悬浮液,加入盛有300mg mar@poly(tapa)的烧杯中,保持250rpm的转速,常温下在365nm紫外灯的照射下反应2h。之后将产物用乙醇和去离子水分别洗涤3次,40℃干燥12h后得到接枝有碳点的双功能大孔吸附树脂材料。其为mar@poly(tapa)-cd微球材料,且微球的粒径为5~10μm,平均粒径为6μm。
[0109]
对实施例1得到的材料进行表征,具体如下:
[0110]
1.材料表征
[0111]
图2:利用扫描电镜sem对合成的大孔吸附树脂进行微观形貌表征,原始mar和改良的mar@poly(tapa)-cd的sem图像如图2所示。单分散的 mar具有均匀的形貌和尺寸分布,平均尺寸约为5μm。如图2a、b、c所示,原始mar为10000、30000、80000时的不同放大倍数,mar表面孔较大,更有利于改性吸附。图2d和e给出了在10000倍和30000倍放大倍数下 mar@poly(tapa)-cd的形貌,与原始mar相似。与原始mar相比, mar@poly(tapa)-cd的表面略有粗糙,可能是由于表面改性。
[0112]
图3:x-射线光电子能谱,利用xps分析了mar@poly(tapa)-cd的化学结构。mar@poly(tapa)-cd的xps全谱如图3a所示。其中c1s、o1s和 si 2p的峰清晰可见,在102ev处的si 2p峰是由功能单体3-(三烯丙基硅基)丙烯酸丙酯引入。此外,在164和400ev处的两个弱峰分别对应于s 2p和n1s 的峰值,其来源于合成的碳点悬浮液。mar@poly(tapa)-cd的n1s、s 2p 和si 2p的高分辨xps光谱如图3b、c和d所示。在n1s的谱图中,结合能在400.1ev处对应的是c-n的出峰位置,在398.0ev处对应为n-h键的出峰。如图3c所示,结合能在100.0和101.5ev处分别对应的是si 2p1/2和2p3/2 的出峰位置,在163.6和166.8ev处分别对应s 2p3/2和2p1/2的出峰。以上结果说明了功能单体3-(三烯丙基硅基)丙烯酸丙酯得到了成功的接枝,并且碳点材料也被成功的固定在了合成的大孔吸附树脂的表面,从而证明了双功能材料 mar@poly(tapa)-cd得到了成功的应用。
[0113]
将实施例1得到的材料用于检测水质中的fe
3+
离子
[0114]
利用双功能材料mar@poly(tapa)-cd对fe
3+
离子的选择性。将浓度为 100μmol l-1
的13种金属离子cu
2+
、cd
2+
、co
2+
、zr4+、pb
2+
、cr
3+
、na+、 k+、li+、fe
3+
、mg
2+
、zn
2+
、ca
2+
与mar@poly(tapa)-cd悬液混合,混合后双功能材料的浓度为0.02mg ml-1
。通过荧光光谱测定每组混合样的荧光强度,不同金属离子与荧光强度之间的猝灭程度如图4a所示。荧光强度随不同金属离子的加入而呈现不同的荧光猝灭程度。在激发波长为360nm的条件下,响应波长在437nm处,加入mg
2+
和zn
2+
离子的强度略有增加;与空白组相比,加入pb
2+
离子后的荧光强度
基本保持不变,而对ca
2+
、cr
3+
、li+、na+、zr4+、 k+等6种金属离子的猝灭度均呈轻微下降趋势。其中4种金属离子cd
2+
、co
3+
、 cu
2+
和fe
3+
对荧光材料的猝灭程度较高,分别达到了31.45%、32.11%、38.61%和46.16%。可见,在添加的13种金属离子中,当加入fe
3+
离子时荧光猝灭效率最高。因此,我们进一步利用fe
3+
离子对双功能材料mar@poly(tapa)-cd 进行定量分析。如图4c所示,双功能材料mar@poly(tapa)-cd在10~80nmoll-1
的线性范围内对fe
3+
离子的具备较好的响应效果。随着fe
3+
离子浓度的增加,437nm处的荧光强度逐渐降低。得到满意的相关系数r=0.9909(图4d)。
[0115]
将实施例1得到的材料用于水质中fe
3+
离子的吸附
[0116]
本发明得到的双功能材料mar@poly(tapa)-cd材料作为一种优良的吸附剂,大孔吸附树脂在吸附和分离方面得到了广泛的应用。本实施例将纳米级 cds接枝到大孔吸附树脂的表面制备得到了双功能材料 mar@poly(tapa)-cd,该材料除了用于检测外,还可作为吸附剂分离水溶液中的fe
3+
离子。
[0117]
首先,静态吸附实验在25℃下吸附5h,之后测定吸附后上清液中的fe
3+
浓度,从而绘制如图5a所示的等温吸附曲线,从图中可以看出,双功能材料的吸附量从25~100μmol l-1
之内迅速增加,当fe
3+
离子浓度为200μmol l-1
时,其最大吸附量达到32.9mg g-1
。并且将得到的数据采用langmuir和 freundlich模型进行拟合,如图5b和5c所示,其线性相关系数分别为0.9909 和0.7748。这一结果说明,该吸附过程更符合langmuir模型,进一步说明该吸附过程为单分子层吸附。综上所述,本发明所涉及的双功能材料,可以同时应用于水质中fe
3+
离子的吸附与检测,具备极好的性能。另外由于本发明整个制备过程简单、反应条件温和、原材料易得价廉,因此适合于大规模制备。
[0118]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1