一种用于监控及纠正测序污染的分子编码检测体系及其应用的制作方法
1.本发明属于基因测序技术领域,涉及一种用于监控及纠正测序污染的分子编码检测体系及其应用。
背景技术:
2.二代测序技术(ngs)因其超大的信息通量,样本容量和超高的灵敏度以及同时检测多分析目标的能力及单样本分析成本低的优势,成为了现代生物学研究和医学诊断的新兴技术。基于ngs技术的诊断产品越来越多的获得医药监管部门的批准,实现了商业化和技术标准化、工业化,但因为ngs技术流程长、过程复杂、批量建库、集中检测带来的样本间污染问题也造成了工业化诊断的隐患。
3.ngs检测污染一般有三个来源:(1)样本处理污染,包括样本信息错误,样本采集过程及核酸提取过程中出现的交叉污染;(2)检测过程污染,一般为复杂的建库过程中出现的试剂如接头index污染或者建库中间产物间的携带或者交叉污染,在同批次大量样本同步建库过程中尤为常见;(3)检测环境污染,由检测环境中的高浓度气溶胶污染分子导致。
4.现有的集中上机测序的pooling方法是用分子标签进行文库标记,即用带有额外文库识别序列信息的接头或者引物进行独立建库,通过下机后数据的标签信息回溯分离样本数据,这种做法只是多样本库同时测序信息再分离的解决方案,并不能识别对样本在样本后处理和建库过程的误操作和交叉污染或者残留污染。任何在pooling过程中的污染都会被携带进入测序流程,而这种污染本身也无法通过从下机后数据质控识别和预处理,只能在数据结果分析后才能察觉某个样本是否出现了操作过程污染。样本标签试剂本身在建库过程中的交叉污染甚至会造成人工假污染,即数据污染。现有的样本污染识别、监控的方法主要是通过被动的分析病人样本的性别、对照样本和检测样本的遗传snp一致度、杂和度等方式实行,必须要等到分析结束后才能得到是否污染的信息,污染后也无法回溯污染来源。对于无对照样本,或者小的靶向测序panel则无法实施。ngs工业化检测急需一种全新的体系来解决上述的样本污染问题。
5.综上所述,如何提供一种能够用于监控、识别及纠正高通量测序样本污染的方法,对于基因测序技术领域具有重要意义。
技术实现要素:
6.针对现有技术的不足和实际需求,本发明提供一种用于监控及纠正测序污染的分子编码检测体系及其应用,本发明设计插入式分子内标记及其回收系统,对待测样本进行标记,通过对下机数据进行分析前特定的编码序列分析,能够及时有效的识别短期内批内样本间交叉污染和由于长期批次检测造成的历史环境污染。
7.为达上述目的,本发明采用以下技术方案:
8.第一方面,本发明提供一种用于监控及纠正测序污染的分子编码检测体系,所述分子编码检测体系包括至少一条插入式编码核酸序列,所述插入式编码核酸序列包括序列
已知的骨架序列区和至少一个可变编码区,所述可变编码区为由a、t、c或g中任意一种或至少两种组成的随机序列,所述可变编码区随机分布于所述骨架序列区内,所述插入式编码核酸序列为单链或双链。
9.本发明中,设计特定结构的插入式编码核酸序列,一部分为固定的已知参考骨架序列,用于信息回收时的序列回帖比对,另一部分为可变编码区,用于特定的样本信息编码,以进行污染识别。利用所述插入式编码核酸序列对待测样本进行标记,基于高通量测序原始数据进行分析,即可快速有效、识别短期内批内样本间交叉污染和由于长期批次检测造成的历史环境污染,可以作为一套标准的ngs试剂就进行检测实验室质量评估和无复检样本污染检测结果的清洗矫正补救。
10.本发明中,选择序列已知的序列为骨架序列区,保证与待测样本无同源性。
11.优选地,所述插入式编码核酸序列的长度为100~2000bp,包括但不限于101bp、102bp、103bp、104bp、105bp、120bp、200bp、220bp、240bp、260bp、280bp、300bp、500bp、800bp、1000bp、1200bp、1300bp、1400bp、1600bp、1700bp、1800bp、1900bp、1950bp、1980bp、1990bp、1995bp、1998bp或1999bp,优选为200~300bp。
12.优选地,所述可变编码区的长度为1~20bp,包括但不限于2bp、3bp、4bp、5bp、6bp、7bp、8bp、10bp、12bp、15bp、16bp、17bp、18bp或19bp,个数为1~4个。
13.优选地,所述插入式编码核酸序列根据所述可变编码区不同分为用于识别批间污染的插入式编码核酸序列或用于识别批内污染的插入式编码核酸序列。
14.优选地,所述用于识别批间污染的插入式编码核酸序列中可变编码区的长度与所述用于识别批内污染的插入式编码核酸序列中可变编码区的长度不同。
15.本发明中,所述用于识别批内污染的插入式编码核酸序列的长度可根据需求设计。可以为100到2000个碱基,优选的为200~300个碱基,进一步优选的为240个碱基,每一个可变编码区的总长度一般为1到4个碱基。分布于1到4个位置,优选的每个编码区长度为1个碱基,分布于核酸序列的4个位置。
16.本发明中,所述用于识别批间污染的插入式编码核酸序列的可变编码区长度可以为1~20个碱基,优选为5个碱基,优选的,用于识别批间污染的分子编码与用于识别批内样本污染的编码区域和方式不同,如批间识别序列的可变编码为连续碱基区,进一步优选的,为防止测序错误或者测序深度不均一造成的信号噪音或者信息损失,批间识别序列的可变编码区可以为两个独立但编码完全相同的连续碱基区在编码信息提取时增加过滤条件提高信息可信度。
17.优选地,所述用于识别批间污染的插入式编码核酸序列中可变编码区的长度为1bp,个数为4个。
18.优选地,所述用于识别批内污染的插入式编码核酸序列中可变编码区的长度为5bp,个数为2个。
19.优选地,所述用于识别批间污染的插入式编码核酸序列包括seq id no.1所示的序列。
20.优选地,所述用于识别批内污染的插入式编码核酸序列包括seq id no.2所示的序列。
21.seq id no.1:
22.ctaaatcgggggctccctttagggttccgatttagtgctttacggcacctcgactccnnnnacttgattagggtgatggttcacgtagtgggccatcgccctgatagacggtttttcgccctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacnnnnaaccctatctcggtctattcttttgatttataagggattttgccgatttcggcctatt。
23.seq id no.2:
24.cgtggctggccacgacgggcgttccttgcgcagctgtgctcgacgttgncactgaagcgggaagggactggctgctattgggcgaagtgccggggcangatctcctgtcatcccaccttgctcctgccgagaaagtatccatcatgnctgatgcaatgcggcggctgcatacgcttgatccggctacctgcccatncgaccaccaagcgaaacatcgcatcgagcgagcacgtactcgga。
25.其中,n为a、t、c、g中任一个。
26.优选地,所述分子编码检测体系中还包括编码信息回收体系。
27.优选地,所述编码信息回收体系包括与所述插入式编码核酸序列互补的探针或引物。
28.本发明中,根据不同的应用场景,编码信息回收体系可以依据液相杂交捕获或扩增子引物扩增等不同方式实现。在某些应用实例中,在杂交捕获探针库中增加插入式编码核酸序列特异性回收探针库,探针由插入式编码核酸序列的匹配碱基构成,在可变编码区,优选的,由兼并互补序列组成。探针的长度可以为50到200个碱基之间,优选的为120个碱基,探针的数量可以为1条到1000条内的任何数目。回收探针的工作浓度可以为0.1nm~10nm。本发明所述的回收探针的特征在于在探针上有一个到多个生物素(biotin)标记,便于进行回收,具体的,有打断步骤探针富集建库插入式编码核酸序列及回收系统工作示意图如图1所示,基因组dna和插入式编码核酸序列101在超声打断后形成长度约150~200bp的片段102;末端修复后两端加上建库接头形成扩增前文库103;在进行液相杂交捕获过程中,插入式编码核酸序列片段和含有目标序列的基因组片段分别与编码核酸序列回收探针和基因特异性探针结合104,完成捕获。无打断步骤探针富集建库插入式编码核酸序列及回收系统工作示意图如图2所示,部分测序样本底物类型如ctdna进行测序无需进行打断步骤。部分底dna含有目标序列,与编码核酸序列混合201,通过接头连接后形成扩增前,文库202,在进行液相杂交捕获过程中,插入式编码核酸序列片段和含有目标序列的基因组片段分别与编码核酸序列回收探针和基因特异性探针结合203,完成捕获。
29.在另外一些应用实例中,回收体系为与掺入插入式编码核酸序列的5’末端及3’末端10~30个碱基相匹配的引物构成,优选的引物长度为18~25个碱基,回收引物的工作浓度可以为0.1μm~10μm,具体的,扩增子富集建库插入式编码核酸序列及回收系统工作示意图如图3所示,含有目标序列的基因组dna与编码核酸序列混合301,第一轮pcr,5’端带有通用序列和测序引物序列修饰的目标基因特异性引物对以及插入式编码核酸序列回收引物对分别与基因组片段及编码核酸序列结合302,第二轮pcr由5’端分别带有p5、p7、index序列,3’端为于第一轮pcr5’端通用序列匹配的序列组成的引物进行扩增,完成建库303。
30.优选地,所述探针的长度可以为50~200bp,数量为1~1000。
31.优选地,所述引物的长度为18~25bp。
32.优选地,所述用于识别批间污染的插入式编码核酸序列的探针的核酸序列选自seq id no.3和/或seq id no.4所示的序列。
33.seq id no.3:
34.ctaaatcgggggctccctttagggttccgatttagtgctttacggcacctcgactccnnnnacttgattagggtgatggttcacgtagtgggccatcgccctgatagacggtttttcgcc-biotin。
35.seq id no.4:
36.ctttgacgttggagtccacgttctttaatagtggactcttgttccaaactggaacaacnnnnaaccctatctcggtctattcttttgatttataagggattttgccgatttcggcctatt-biotin。
37.优选地,所述用于识别批内污染的插入式编码核酸序列的探针的核酸序列选自seq id no.5和/或seq id no.6所示的序列。
38.seq id no.5:
39.cgtggctggccacgacgggcgttccttgcgcagctgtgctcgacgttgncactgaagcgggaagggactggctgctattgggcgaagtgccggggcangatctcctgtcatcccaccttg-biotin。
40.seq id no.6:
41.ctcctgccgagaaagtatccatcatgnctgatgcaatgcggcggctgcatacgcttgatccggctacctgcccatncgaccaccaagcgaaacatcgcatcgagcgagcacgtactcgga-biotin。
42.优选地,所述用于识别批间污染的插入式编码核酸序列的引物的核酸序列包括seq id no.7和seq id no.8所示的序列。
43.seq id no.7:
44.tcgtcggcagcgtcagatgtgtataagagacagctaaatcgggggctccctttagg。
45.seq id no.8:
46.gtctcgtgggctcggagatgtgtataagagacagaataggccgaaatcggcaaaatccct。
47.优选地,所述用于识别批内污染的插入式编码核酸序列的引物的核酸序列包括seq id no.9和seq id no.10所示的序列。
48.seq id no.9:
49.tcgtcggcagcgtcagatgtgtataagagacagcgtggctggccacgacgggcgttcctt。
50.seq id no.10:
51.gtctcgtgggctcggagatgtgtataagagacagtccgagtacgtgctcgctcgatgcga。
52.本发明中,用于监控及纠正测序污染的分子编码检测体系的应用可以贯穿高通量测序的整个过程如从样本核酸提取时及开始,因此可以将插入式编码核酸序列预制到核酸容器中,用量可以为样本分子数量的1%~1000%,优选为10%~100%。
53.第二方面,本发明提供第一方面所述的用于监控及纠正测序污染的分子编码检测体系在制备基因测序产品中的应用。
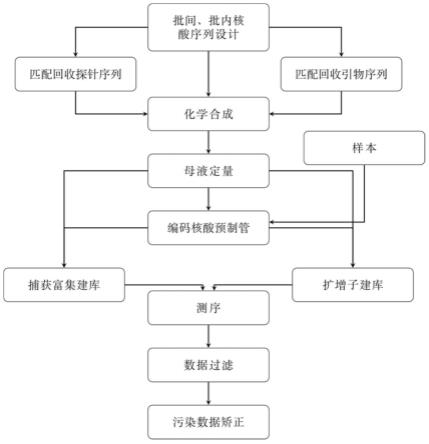
54.本发明设计的用于监控及纠正测序污染的分子编码检测体系可有效应用于制备测序产品,作为监控及纠正测序污染组分。
55.第三方面,本发明提供一种测序试剂盒,所述测序试剂盒包括第一方面所述的用于监控及纠正测序污染的分子编码检测体系。
56.第四方面,本发明提供第一方面所述的用于监控及纠正测序污染的分子编码检测体系在基因测序中应用。
57.第五方面,本发明提供一种监控及纠正测序污染的方法,所述方法包括:
58.将第一方面所述的用于监控及纠正测序污染的分子编码检测体系与待测样品混
合,进行文库构建和纯化,对纯化后文库进行测序,根据测序结果进行数据分析和污染纠正。
59.污染的判断标准为:所述插入式编码核酸序列中所有可变编码区出现非本样本唯一编码序列,并且疑似污染序列的reads数超过3条。
60.所述污染纠正包括:回溯污染指向的样本,进行对照比对,去除本样本假阳性突变。
61.本发明中,监控及纠正测序污染的方法实施流程图如图4所示,分析过程包括将测序数据mapping至参考基因组和参考编码核酸序列,对回收的插入式编码核酸序列进行过滤和有效深度统计,插入式编码核酸序列数据过滤及污染识别的步骤为:数据回帖,批间及批内可变编码区序列提取,重复序列去除和污染识别,污染识别的条件为:
62.(1)批间污染,批间插入式编码核酸序列中可变编码区出现非本样本唯一编码序列,并且疑似污染编码同时存在于所有可变编码区,并且疑似污染序列的reads数超过3条;
63.(2)批内污染,(a)批内插入式编码核酸序列中所有可变编码区同时出现非样本唯一编码序列,并且疑似污染编码序列reads在每一个编码区都超过3条,(b)疑似污染编码序列可以在本批次样本内溯源。
64.有效深度统计(污染指数统计)为目的样本唯一编码有效深度占总回收编码有效深度占比。
65.与现有技术相比,本发明具有以下有益效果:
66.本发明中,设计特定结构的插入式编码核酸序列,利用所述插入式编码核酸序列对待测样本进行标记,基于高通量测序原始数据进行分析,即可快速有效、识别短期内批内样本间交叉污染和由于长期批次检测造成的历史环境污染,可以作为一套标准的ngs试剂就进行检测实验室质量评估和无复检样本污染检测结果的清洗矫正补救。
附图说明
67.图1为有打断步骤探针富集建库插入式编码核酸序列及回收系统工作示意图;
68.图2为无打断步骤探针富集建库插入式编码核酸序列及回收系统工作示意图;
69.图3为扩增子富集建库插入式编码核酸序列及回收系统工作示意图;
70.图4为监控及纠正测序污染的方法实施流程图;
71.图5为本发明用于监控及纠正测序污染的分子编码检测体系性能验证结果图。
具体实施方式
72.为进一步阐述本发明所采取的技术手段及其效果,以下结合实施例和附图对本发明作进一步地说明。可以理解的是,此处所描述的具体实施方式仅仅用于解释本发明,而非对本发明的限定。
73.实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购获得的常规产品。
74.实施例1
75.本实施例设计用于监控及纠正测序污染的分子编码检测体系。
76.一、设计插入式编码核酸序列
77.插入式编码核酸序列为双链dna,骨架序列设计采用外源人工序列。用于识别批间污染的插入式编码核酸序列如seq id no.1所示,由240个碱基组成,blast搜索未与人基因组有任何同源性,其中第58到第61个碱基和179到182个碱基为可变编码区,由nnnn表示,每个n表示a、t、c、g中的任意一个,第58到第61个碱基和第179个到182个碱基的序列完全相同,根据n不同组合,设计256个不同的插入式编码核酸序列,其捕获回收探针序列为单链dna,由两条120个碱基序列组成,分别匹配seq id no.1中1到120个碱基,和121-240个碱基,其中匹配可变编码区序列为兼并序列,捕获探针序列在3’有一个碱基具有生物素标记,具体序列如seq id no.3和seq id no.4所示,插入式编码核酸序列的扩增子回收引物由正反向两条引物构成,分别对应seq id no.1的5’端23个碱基和3’端26个碱基,其中每条引物的5’分别带有建库引物匹配的融合序列,具体序列如seq id no.7和seq id no.8所示。
78.用于识别批内污染的插入式编码核酸序列如seq id no.2所示,由240个碱基组成,blast搜索未与人基因组有任何同源性,其中第49、98、147、196个碱基为可变编码区n,第49个碱基编码为a、t、c或g,第98个碱基编码为t或c,第147个碱基编码为a、c或g,第196个碱基编码为a、t、c或g。根据n不同组合,设计96个不同的插入式编码核酸序列,其捕获回收探针序列为单链dna,由两条120个碱基序列组成,分别匹配seq id no.2中1到120个碱基和121到240个碱基,其中可变编码区为简并碱基,3’端带有生物素标记,具体序列如seq id no.5和seq id no.6所示,其扩增子回收引物由正反向两条引物构成,分别对应seq id no.2中5’端27个碱基和3’端26个碱基,其中每条引物的5’端分别带有建库引物匹配融合序列,具体序列如seq id no.9和seq id no.10所示。
79.二、化学合成
80.将设计的256条用于识别批间污染的插入式编码核酸序列和匹配的捕获回收探针及回收引物序列以及96条用于识别批内污染的插入式编码核酸序列和匹配的捕获回收探针及回收引物序列委托合成(integrated dna technologies),合成序列形式为干粉。
81.母液配制及定量按照合成产品说明书将掺入式双链核酸序列及其匹配回收探针及引物加入超纯水配置成100μm母液,然后批间、批内掺入式双链核酸继续稀释到按照30000copies/μl的预制液生产浓度,将2μl预制液加入1.5ml ep管底,将预制管置入-80℃冰箱保存。
82.实施例2
83.本实施例进行有打断捕获富集建库(靶向panel)使用测试,其工作原理如图1所示,具体包括以下步骤:
84.(1)样本dna提取,所测试样本类型为ffpe样本,样本dna提取试剂盒为qiaamp dna ffpe tissue kit,提取后dna定量后取200ng加入预制管,其中预制管的识别批间污染的插入式编码核酸序列的可变编码区的碱基为aggt,识别批内污染的插入式编码核酸序列的可变编码区的碱基按照5’到3’的顺序分别为a、t、c、c,提取dna加入预制管后轻微涡旋震荡30s;
85.(2)建库步骤,本实施案例中所涉及建库试剂采购自neb,探针杂交试剂来自idt;
86.a.核酸打断步骤,将dna用1
×
te buffer补充体积到50μl,利用covaris m220超声打断仪按照以表l步骤进行dna打断;
87.表1
88.占空比10%峰值功率75突发循环数200打断时长100-330s水浴温度18-20℃
89.b.打断修复,按表2配置反应液,20℃孵育15min;
90.表2
91.片段化ffpe dna50μlffpe dna缓冲液6.5μlnebnext ffpe dna repair mix2μl超纯水3.5μl总共62μl
92.c.磁珠纯化及末端修复利用ampure xp beads进行核酸纯化,利用nebnext ultra ii end prep kit进行末端修复,修复反应体系及pcr程序如表3和表4所示;
93.表3
94.ffpe dna50μlnebnext ultra
ꢀⅱꢀ
end prep buffer7μlnebnext ultra
ꢀⅱꢀ
end prep enzyme mix3μl总共60μl
95.表4
96.步骤温度时间循环120℃30min循环265℃30min循环34℃暂停
97.d.接头连接,按照表5反应体系进行建库接头连接,20℃,孵育15min;
98.表5
99.dna repair reaction mixture60μlnebnext ultra
ꢀⅱꢀ
ligation master mix30μlnebnext ligation enhancer1μlduplex seq adapters2μl总共93μl
100.e.文库片段筛选及预扩增,ampure xp beads进行片段筛选,然后按照表6反应体系及表7反应条件进行接头预扩增;
101.表6
102.nebnext ultra
ꢀⅱꢀ
q5 master mix25μludi primer mix5μl总共30μl
103.表7
[0104][0105]
f.杂交捕获,按照表8反应体系及表9反应条件进行靶向序列及编码核酸序列捕获;
[0106]
表8
[0107]
2x hybridization buffer8.5μlhybridization buffer enhancer2.7μl靶向基因panel4μl批间批内编码核酸回收探针1.8μl总共17μl
[0108]
表9
[0109]
步骤温度时间循环195℃30s循环265℃4h循环365℃暂停
[0110]
g.链霉素磁珠回收、捕获文库扩增及纯化,参照dynabeads m-270使用说明书回收洗涤杂交捕获序列,并按照表10反应体系及表11反应条件进行捕获文库扩增;
[0111]
表10
[0112]
library pcr master mix(2
×
)25μlillumina p5/p7 primer mix(10
×
)5μldynabeads20μl总共50μl
[0113]
表11
[0114][0115]
h.文库纯化及定量
[0116]
利用ampure xp beads进行扩增后文库纯化,利用qubit 3.0进行纯化后文库定量。
[0117]
(3)测序
[0118]
利用novaseq 6000高通量测序仪pe150读长进行上机测序,测序深度为10000
×
,数据mapping至参考基因组和参考编码核酸序列,对回收的插入式编码核酸序列进行过滤和有效深度统计,插入式编码核酸数据过滤及污染判别的标准和步骤为:数据回帖,批间及批内可变编码区序列提取,重复序列去除,污染识别,污染识别的条件为:1)批间污染,批间插入式编码序列可变编码区出现非本样本唯一编码序列,并且疑似污染编码同时存在于第一可变编码区和第二可变编码区;并且疑似污染序列的reads数超过3条;2)批内污染,(a)批内插入式编码第1、2、3、4可变编码区同时出现非样本唯一编码序列,并且疑似污染编码序列reads在每一个可变编码区都超过3条,(b)疑似污染编码序列可以在本批次样本内溯源。
[0119]
污染指数统计:目的样本唯一编码有效深度占总回收编码有效深度占比。
[0120]
污染纠正:回溯批间污染及批内污染指向的样本,通过对照比对,去除本样本假阳性突变。
[0121]
本次实施的批间、批内插入式编码序深度如表12所示,证明该方法可以回收足够数量的样本唯一识别编码。
[0122]
表12
[0123]
测序目标有效深度5192
×
批间插入式编码序列有效深度4567
×
批内插入式编码序有效深度4605
×
[0124]
实施例3
[0125]
本实施例进行扩增子建库方法(trb免疫组库靶向测序),其工作原理如图3所示,包括以下步骤:
[0126]
1、样本dna提取,所测试样本类型为血液样本,样本dna提取试剂盒为qiaamp dna blood kit.提取后dna定量后取1μg加入预制管,其中预制管的批间插入式编码核酸序列可
变编码区的碱基为atat,批内插入式编码核酸序列可变编码区的的碱基按照5’到3’的顺序分别为t、t、c、t,提取dna加入预制管后轻微涡旋震荡30s;
[0127]
2、pcr扩增富集目的及编码序列,使用trb引物体系多重pcr扩增目标区域,按表13配置反应体系,所涉及的批内、批间核酸引物对seq id no.7、seq id no.8、seq id no.9和seq id no.10所示,反应条件如表14所示;
[0128]
表13
[0129]2×
multiplex pcr buffer25μlmultiplex polymerase1μltrb primer mix(10μm)2μl批间编码核酸引物工作液2μl批内编码核酸引物工作液2μl超纯水2μldna(1000ng)20μl
[0130]
表14
[0131][0132]
3、利用ampure xp beads进行纯化后进行建库pcr按表15反应体系(其中p5-f及p7-r序列如seq id no.11(aatgatacggcgaccaccgagatctacacatatgcgctcgtcggcagcgtc)和seq id no.12(caagcagaagacggcatacgagatctgatcgtgtctcgtgggctcgg)所示)及表16反应条件进行;
[0133]
表15
[0134]
5x reaction buffer10μldna polymerase0.5μl10mm dntp1μlp5-f(10um)1μlp7-r(10um)1μlnuclease-free water34.5μl
[0135]
表16
[0136][0137]
4、文库纯化及测序
[0138]
利用ampure xp beads进行扩增后文库纯化,利用qubit 3.0进行纯化后文库定量,novaseq 6000高通量测序仪pe150读长进行上机测序,测序量为0.3mreads.
[0139]
5、数据分析及深度统计
[0140]
数据分析及深度统计方法按照实施案例1所述,本次实施的批间、批内插入式编码序列深度如表17所示,证明扩增子建库方法可以回收足够数量的样本唯一识别编码。
[0141]
表17
[0142]
批间编码有效reads63425批内编码有效reads52583
[0143]
实施例4
[0144]
本实施例进行人工混合样本的污染识别性能验证。
[0145]
分别制备人工模拟掺比污染样本进行污染识别能力的性能验证,污染掺比的比例梯度为0.1%、0.5%、l%、5%和10%,实际污染指数的数据如图5所示,证明本发明用于监控及纠正测序污染的分子编码检测体系可以最低识别0.1%水平的污染。
[0146]
综上所述,本发明设计特定结构的插入式编码核酸序列,利用所述插入式编码核酸序列对待测样本进行标记,基于高通量测序原始数据进行分析,即可快速有效、识别短期内批内样本间交叉污染和由于长期批次检测造成的历史环境污染,可以作为一套标准的ngs试剂就进行检测实验室质量评估和无复检样本污染检测结果的清洗矫正补救。
[0147]
申请人声明,本发明通过上述实施例来说明本发明的详细方法,但本发明并不局限于上述详细方法,即不意味着本发明必须依赖上述详细方法才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1