一种一维链状Cu(Ⅱ)配合物及其制备方法和催化转化二氧化碳的应用
一种一维链状cu(ⅱ)配合物及其制备方法和催化转化二氧化碳的应用
技术领域
1.本发明涉及二氧化碳催化转化技术领域,具体涉及一种一维链状cu(ⅱ)配合物及其制备方法和催化转化二氧化碳的应用。
背景技术:
2.随着温室效应日益加重,如何有效处理大气中的二氧化碳迫在眉睫。对二氧化碳的固定存储以及增值转化是处理二氧化碳的有效方式,在各种二氧化碳转换反应中,通过筛选不同环氧化合物作为底物,与二氧化碳发生环加成反应以达到固定和循环大气中二氧化碳的目的,将其转化为有价值的化学品,如在医药和精细化工领域有着广泛应用的环状碳酸酯,是一种原子利用率最高且很有前景的方法。
3.对于二氧化碳与环氧化合物环加成反应而言,已报道出系列催化剂,包括金属氧化物、碱金属盐、salen金属配合物、季铵/或鏻盐、金属有机骨架化合物以及离子液体。然而,报道的大部分催化剂存在以下弊端:(1)催化剂合成成本高、合成过程复杂;(2)催化反应条件苛刻(高温、高压)、助催化剂易流失、使用挥发性有机溶剂(n,n-二甲基甲酰胺、氯苯、甲基乙基酮)等,并不适合工业应用。因此,开发高活性催化剂成为必然的发展趋势。
技术实现要素:
4.本发明的发明目的在于:针对上述存在的问题,提供一种一维链状cu(ⅱ)配合物及其制备方法和催化转化二氧化碳的应用,本发明的催化剂具有较高的催化活性,在常压、80℃即可催化转化二氧化碳,产率可达99%。
5.为了实现上述目的,本发明采用的技术方案如下:
6.一种一维链状cu(ⅱ)配合物,其化学通式为[cu(l5)2(py)2]n,其中,py为吡啶,l5为失去一个氢离子的羧酸配体,l5的结构式如下:
[0007][0008]
所述配合物的金属中心铜离子为正二价,其为四配位,与源自于2个l5配体的氧原子和2个吡啶配体的氮原子相连,继而铜与铜之间通过一个l5配体桥联为一维螺旋链状。
[0009]
本发明中,优选地,所述配合物结晶于三斜晶系,p1空间群,晶胞参数为:本发明中,优选地,所述配合物结晶于三斜晶系,p1空间群,晶胞参数为:α=90
°
,β=90
°
,γ=120
°
;
[0010]
本发明还提供上述一维链状cu(ⅱ)配合物的制备方法,包括以下步骤:
[0011]
(1)配体l5的合成;
[0012]
(2)一维链状cu(ⅱ)配合物的合成:将配体l5、cucl2·
2h2o与混合溶剂按照一定比例混合,在75-85℃的条件下反应,冷却后析出蓝色晶体,即得;其中,所述混合溶剂中dmf、h2o、meoh、1m hcl和吡啶的体积比为10:3:3:5:1;配体l5与cucl2·
2h2o、混合溶剂按照mol/mol/ml比计为3:4:180-260。
[0013]
上述制备方法中,优选地,所述配体l5的合成步骤如下:以1,1-联-2-萘酚为l1原料,将l1与溴乙烷反应得到l2,l2与液溴反应得到l3,l3与cucn反应得到l4,l4与碱反应得到l5。
[0014]
上述制备方法中,优选地,所述配体l5的合成步骤如下:
[0015]
(1)制备l2:将l1、k2co3、溴乙烷和乙腈按照mol/mol/mol/ml比计为1:3:6:300-500混合在反应容器中,混合物在75-85℃回流反应2天,用乙酸乙酯萃取干燥,得l2白色粉末;其中,l1为1,1-联-2-萘酚;
[0016]
(2)制备l3:取l2于反应容器中,加入100ml冰醋酸,l2与冰醋酸按mmol/ml比计为1:5;液溴用冰醋酸稀释后在常温下滴加,反应10-30小时后,将冰醋酸去除,萃取干燥,得到l3白色粉末;
[0017]
(3)制备l4:取l3于反应容器中,加入cucn、dmf,l3与cucn按摩尔比计为1:10,在氮气保护下,在150-160℃温度条件下反应回流36-60小时,待反应冷却后加入氨水,过滤,过柱,提纯得l4白色粉末;
[0018]
(4)制备l5:取l4于反应容器中,用6m氢氧化钠溶液和等量乙醇溶液溶解,l4与氢氧化钠溶液、乙醇溶液按mmol/ml/ml比计为14:100:100;在75-85℃条件下反应回流10-20小时,待反应冷却后,加入浓hcl,调节ph至0.9-1.1,用乙酸乙酯萃取,无水mgcl2干燥得l5白色粉末。
[0019]
上述制备方法中,优选地,步骤(2)中所述的反应时间为3-5天。
[0020]
本发明还提供上述一维链状cu(ⅱ)配合物在催化转化二氧化碳中的应用,也即上述制备方法制备得到的一维链状cu(ⅱ)配合物在催化转化二氧化碳中的应用。
[0021]
具体地,该应用为,使用一维链状cu(ⅱ)配合物作为晶体催化剂,四丁基溴化铵作为助催化剂,使环氧化合物和二氧化碳环通过加成反应合成环状碳酸酯。优选地,所述加成反应的条件为常压、80℃,所述晶体催化剂和助催化剂的用量均为环氧化合物摩尔用量的0.15%,反应时间为1-60小时。
[0022]
综上所述,由于采用了上述技术方案,本发明的有益效果是:
[0023]
1、本发明提供了一维链状cu(ⅱ)配合物,该配合物具有较高的催化转化二氧化碳的活性,能够催化转化二氧化碳与环氧化合物合成环状碳酸酯。该催化剂在催化二氧化碳转化合成环状碳酸酯反应中,反应条件温和(80℃、常压)、催化活性高、选择性好,催化剂易于回收,转化率可达99%。
[0024]
2、本发明还提供一维链状cu(ⅱ)配合物的制备方法,所述制备方法简单易得、合成高效。
附图说明
[0025]
图1为本发明的cu(ii)配合物的流程图。
[0026]
图2为本发明的cu(ii)配合物的结构图。
[0027]
图3为本发明的cu(ii)配合物的合成路线图。
[0028]
图4为本发明实施制得的配体l5和cu(ii)配合物的红外光谱图。
[0029]
图5为本发明实施制得cu(ii)配合物的粉末x-射线衍射图。
具体实施方式
[0030]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0031]
本发明提供一种一维链状cu(ⅱ)配合物,其化学通式为[cu(l5)2(py)2]n,其中,py为吡啶,l5为失去一个氢离子的羧酸配体,l5的结构式如下:
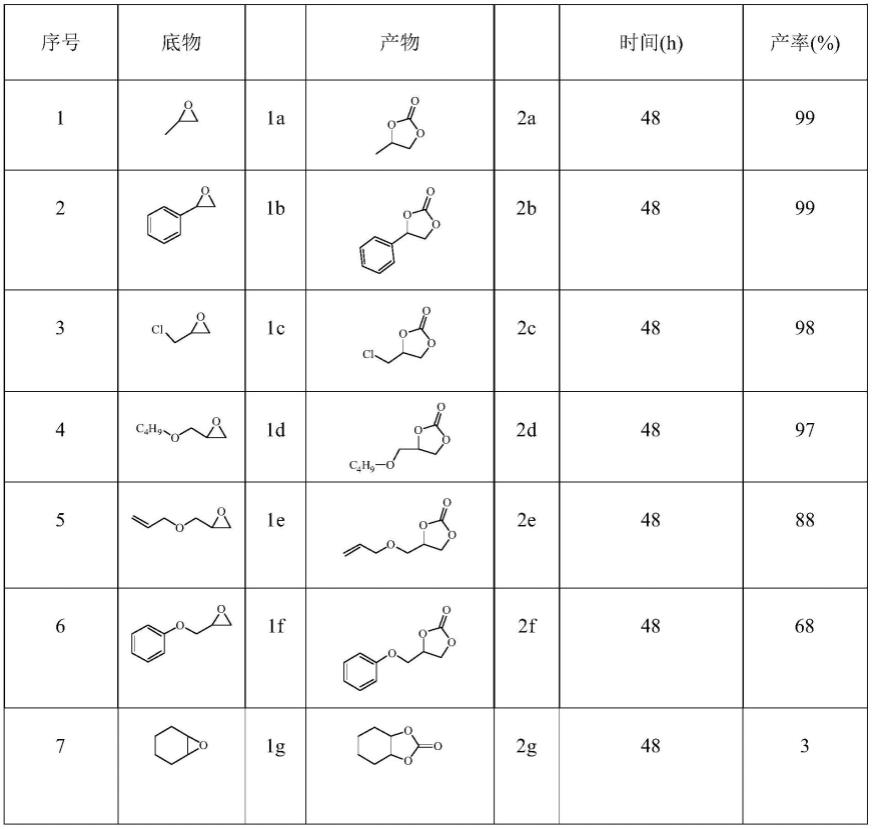
[0032][0033]
其中,配合物的金属中心铜离子为正二价,其为四配位,与源自于2个l5配体的氧原子和2个吡啶配体的氮原子相连,继而铜与铜之间通过一个l5配体桥联为一维螺旋链状。该配合物结晶于三斜晶系,p1空间群,晶胞参数为:斜晶系,p1空间群,晶胞参数为:α=90
°
,β=90
°
,γ=120
°
;参见图1-2,其中,图1为cu(ⅱ)配合物的单元结构图;图2为一维链状cu(ⅱ)配合物的结构图。
[0034]
一维链状cu(ⅱ)配合物的合成路线为:以1,1-联-2-萘酚为l1原料,将l1与溴乙烷反应得到l2,l2与液溴反应得到l3,l3与cucn反应得到l4,l4与碱反应得到l5,l5、cucl2·
2h2o与混合溶剂一起合成最终产物,具体合成路线如图3所示。
[0035]
以下通过具体实施例介绍一维链状cu(ⅱ)配合物的制备方法。
[0036]
实施例1
[0037]
一维链状cu(ⅱ)配合物的制备方法,包括以下步骤
[0038]
(1)配体l5的合成;
[0039]
①
制备l2:将l1、k2co3、溴乙烷和乙腈按照mol/mol/mol/ml比计为1:3:6:300混合在反应容器中,混合物在75℃回流反应2天,用乙酸乙酯萃取干燥,得l2白色粉末;其中,l1为1,1-联-2-萘酚。
[0040]
l2白色粉末的氢谱数据为:1h nmr(400mhz,cdcl3)δ7.93(d,j=9.0hz,1h),7.85(d,j=8.1hz,1h),7.41(d,j=9.0hz,1h),7.30(d,j=8.1,6.6,1.3hz,1h),7.19(d,j=8.0,6.6,1.3hz,1h),7.13(dd,j=8.0,0.5hz,1h),4.13
–
3.92(m,2h),1.05(t,j=7.0hz,3h),证实其结构为合成路线图中的l2。
[0041]
②
制备l3:取l2于反应容器中,加入100ml冰醋酸,l2与冰醋酸按mmol/ml比计为1:
5;液溴用冰醋酸稀释后在常温下滴加,液溴与l2的摩尔比为1:1,反应30小时后,将冰醋酸去除,萃取干燥,得到l3白色粉末。
[0042]
l3白色粉末的氢谱数据为:1h nmr(400mhz,cdcl3)δ8.00(d,j=2.0hz,1h),7.84(d,j=9.0hz,1h),7.45
–
7.39(d,1h),7.29
–
7.22(dd,1h),6.96(d,j=7.2hz,1h),4.11
–
3.96(m,2h),1.05(t,j=7.0,3.5hz,3h),证实其结构为合成路线图中的l3。
[0043]
③
制备l4:取l3于反应容器中,加入cucn、dmf,l3与cucn按摩尔比计为1:10,在氮气保护下,在150℃温度条件下反应回流60小时,待反应冷却后加入氨水,过滤,过柱,提纯得l4白色粉末;
[0044]
l4白色粉末的氢谱数据为:1h nmr(400mhz,cdcl3)δ8.26(d,j=1.5hz,1h),8.02(d,j=9.0hz,1h),7.56
–
7.49(d,1h),7.37
–
7.29(dd,1h),7.12(d,j=8.8hz,1h),4.18
–
4.03(m,2h),1.09(dd,j=8.8,5.2hz,3h),证实其结构为合成路线图中的l4。
[0045]
④
制备l5:取l4于反应容器中,用6m氢氧化钠溶液和等量乙醇溶液溶解,l4与氢氧化钠溶液、乙醇溶液按mmol/ml/ml比计为14:100:100;在75℃条件下反应回流20小时,待反应冷却后,加入浓hcl,调节ph至0.9,用乙酸乙酯萃取,无水mgcl2干燥得l5白色粉末。
[0046]
l5白色粉末的氢谱数据为:1h nmr(400mhz,dmso)δ12.87(s,1h),8.69(d,j=1.5hz,1h),8.32(d,j=9.1hz,1h),7.85
–
7.61(m,2h),7.04(d,j=8.9hz,1h),4.30
–
4.13(m,2h),1.07(t,j=7.0hz,3h),证实其结构为合成路线图中的l5。
[0047]
(2)一维链状cu(ⅱ)配合物的合成:将配体l5、cucl2·
2h2o与混合溶剂按照一定比例混合,在75℃的条件下反应5天,冷却后析出蓝色晶体,即得;其中,所述混合溶剂中dmf、h2o、meoh、1m hcl和吡啶的体积比为10:3:3:5:1;配体l5与cucl2·
2h2o、混合溶剂按照mol/mol/ml比计为3:4:180。
[0048]
实施例2
[0049]
一维链状cu(ⅱ)配合物的制备方法,包括以下步骤:
[0050]
(1)配体l5的合成;
[0051]
①
制备l2:将l1、k2co3、溴乙烷和乙腈按照mol/mol/mol/ml比计为1:3:6:400混合在反应容器中,混合物在80℃回流反应2天,用乙酸乙酯萃取干燥,得l2白色粉末(16.8g,产率:98%);其中,l1为1,1-联-2-萘酚;氢谱数据证实其结构为合成路线图中的l2。
[0052]
②
制备l3:取l2于反应容器中,加入100ml冰醋酸,l2与冰醋酸按mmol/ml比计为1:5;液溴用冰醋酸稀释后在常温下滴加,液溴与l2的摩尔比为1:1,反应20小时后,将冰醋酸去除,萃取干燥,得到l3白色粉末(9.5g,产率:95%);氢谱数据证实其结构为合成路线图中的l3。
[0053]
③
制备l4:取l3于反应容器中,加入cucn、dmf,l3与cucn按摩尔比计为1:10,在氮气保护下,在155℃温度条件下反应回流48小时,待反应冷却后加入氨水,过滤,过柱,提纯得l4白色粉末(2.2g,产率:56%);氢谱数据证实其结构为合成路线图中的l4。
[0054]
④
制备l5:取l4于反应容器中,用6m氢氧化钠溶液和等量乙醇溶液溶解,l4与氢氧化钠溶液、乙醇溶液按mmol/ml/ml比计为14:100:100;在80℃条件下反应回流15小时,待反应冷却后,加入浓hcl,调节ph至1.0,用乙酸乙酯萃取,无水mgcl2干燥得l5白色粉末(2.4g,产率:99%),氢谱数据证实其结构为合成路线图中的l5。
[0055]
(2)一维链状cu(ⅱ)配合物的合成:将配体l5、cucl2·
2h2o与混合溶剂按照一定比
例混合,在80℃的条件下反应4天,冷却后析出蓝色晶体,即得(基于铜产率:45%);其中,所述混合溶剂中dmf、h2o、meoh、1m hcl和吡啶的体积比为10:3:3:5:1;配体l5与cucl2·
2h2o、混合溶剂按照mol/mol/ml比计为3:4:200。
[0056]
实施例3
[0057]
一维链状cu(ⅱ)配合物的制备方法,包括以下步骤
[0058]
(1)配体l5的合成;
[0059]
①
制备l2:将l1、k2co3、溴乙烷和乙腈按照mol/mol/mol/ml比计为1:3:6:500混合在反应容器中,混合物在85℃回流反应2天,用乙酸乙酯萃取干燥,得l2白色粉末;其中,l1为1,1-联-2-萘酚;氢谱数据证实其结构为合成路线图中的l2。
[0060]
②
制备l3:取l2于反应容器中,加入100ml冰醋酸,l2与冰醋酸按mmol/ml比计为1:5;液溴用冰醋酸稀释后在常温下滴加,液溴与l2的摩尔比为1:1,反应10小时后,将冰醋酸去除,萃取干燥,得到l3白色粉末;氢谱数据证实其结构为合成路线图中的l3。
[0061]
③
制备l4:取l3于反应容器中,加入cucn、dmf,l3与cucn按摩尔比计为1:10,在氮气保护下,在160℃温度条件下反应回流36小时,待反应冷却后加入氨水,过滤,过柱,提纯得l4白色粉末;氢谱数据证实其结构为合成路线图中的l4。
[0062]
④
制备l5:取l4于反应容器中,用6m氢氧化钠溶液和等量乙醇溶液溶解,l4与氢氧化钠溶液、乙醇溶液按mmol/ml/ml比计为14:100:100;在85℃条件下反应回流10小时,待反应冷却后,加入浓hcl,调节ph至1.1,用乙酸乙酯萃取,无水mgcl2干燥得l5白色粉末;氢谱数据证实其结构为合成路线图中的l5。
[0063]
(2)一维链状cu(ⅱ)配合物的合成:将配体l5、cucl2·
2h2o与混合溶剂按照一定比例混合,在75℃的条件下反应3天,冷却后析出蓝色晶体,即得;其中,所述混合溶剂中dmf、h2o、meoh、1m hcl和吡啶的体积比为10:3:3:5:1;配体l5与cucl2·
2h2o、混合溶剂按照mol/mol/ml比计为3:4:260。
[0064]
图4为本发明实施制得配体的l5和cu(ii)配合物的红外光谱图;图5为本发明实施例制得cu(ii)配合物的粉末x-射线衍射图,制得晶体所测粉末与模拟所得基本吻合。
[0065]
本发明制备所得到的一维链状cu(ⅱ)配合物具有催化转化二氧化碳中的活性,可以用于催化转化二氧化碳。具体方法如下:
[0066]
使用一维链状cu(ⅱ)配合物作为晶体催化剂,四丁基溴化铵作为助催化剂,使环氧化合物和二氧化碳环通过加成反应合成环状碳酸酯。具体方法为:
[0067]
取20mmol环氧化合物于反应管内,加0.15mol%晶体催化剂,0.15mol%四丁基溴化铵,超声后抽出管内空气,接常压二氧化碳气球,于80℃反应1-60h,离心分离通过核磁确定产率。合成路线如下:
[0068][0069]
以其他环氧化合物为反应物,按上述相同的条件反应两天,计算产率,其结果如下表1所示:
[0070]
表1
[0071][0072]
从表1的数据可以看出,同样的条件下,效果最佳的反应底物为环氧氯丙烷,同时也说明本发明的一维链状cu(ⅱ)配合物具有催化转化二氧化碳中的活性,反应条件温和(80℃、常压)、催化活性高、选择性好,催化剂易于回收,转化率可达99%。
[0073]
上述说明是针对本发明较佳可行实施例的详细说明,但实施例并非用以限定本发明的专利申请范围,凡本发明所提示的技术精神下所完成的同等变化或修饰变更,均应属于本发明所涵盖专利范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1