一种7-脱氢胆固醇中间体的制备方法及7-脱氢胆固醇的制备方法与流程
1.本发明涉及7-脱氢胆固醇合成技术领域,更具体地,涉及一种7-脱氢胆固醇中间体的制备方法及7-脱氢胆固醇的制备方法。
背景技术:
2.维生素d3属于类甾醇衍生物,对于骨骼的形成有着重要的意义。7-脱氢胆固醇是维生素d3的前体,想要得到维生素d3,先要合成出7-脱氢胆固醇,目前7-脱氢胆固醇的合成方法主要有:
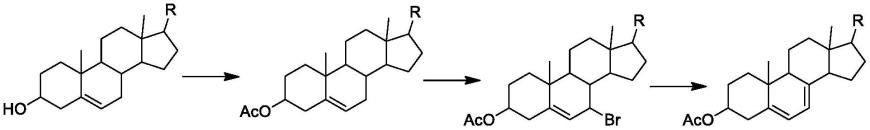
3.一、溴化、脱溴化氢法
4.该方法的工艺路线如下:
[0005][0006]
该工艺路线存在以下缺点:
①
生物提取胆固醇中含副产物,它的侧链双键的α位也会被溴化;
②
溴化氢不易脱去生成相应的共轭二烯;
③
制备7-脱氢胆固醇过程产生的副产物难于分离纯化且收率低;
④
使用的溴对环境有害,且不易除去,不符合绿色化学的要求,而且产量从不高;
⑤
从胆固醇到7-脱氢胆固醇的收率只有40%而且含羞副产物难以去除干净,导致产率降低以及产品杂质多,分离纯化难。
[0007]
二、氧化、还原、消除法
[0008]
该方法的工艺路线如下:
[0009][0010]
该方法避免了老工艺中副产物多和溴试剂污染环境的缺陷,但不足之处在于
①
烯丙位氧化:传统的烯丙位氧化方法和改进的烯丙位方法有很多,但所用氧化剂对环境均造成污染且成本高;
②
羰基还原成羟基过程中加氢为较绿色环保的途径,但氢气易爆炸有一定危险性。
[0011]
三、氧化、成腙、脱腙
[0012]
该方法的工艺路线如下:
[0013][0014]
烯丙位氧化及腙消除反应是这个工艺中最关键也是最难的两步反应,收率不高,原子经济性低,对环境不友好。
[0015]
四、b环通过重排引入5、7双键,侧链偶联(如专利cn112608361a)
[0016]
该方法的工艺路线如下:
[0017][0018]
该方法存在以下缺陷:
①
工艺中使用的硼氢化钠位危险试剂;
②
收率低,从起始物ba到目标产物脱氢胆固醇,最终收率约40%;3、5,7-双键引入时通过酯化、硼氢化钠还原获得3β-羟基-5,7-二烯结构,易产生异构体杂质,该杂质与目标产物类似难以通过简单方法去除,导致纯度与收率不理想。
技术实现要素:
[0019]
基于现有技术中存在的上述技术问题,本发明提供了一种7-脱氢胆固醇中间体的制备方法,通过该方法可提高7-脱氢胆固醇中间体的收率和纯度,进一步提高7-脱氢胆固醇的收率。
[0020]
为了实现上述目的,本发明的技术方案如下:
[0021]
一种制备高纯化合物v的方法,其特征在于,所述化合物v的结构式为:该方法包括以下步骤:
[0022]
向第一反应容器中加入化合物iv、第一溶剂、水、酮还原酶、酯水解酶、辅酶、辅酶
再生系统,在25~35℃下进行反应,得到化合物v;
[0023]
其中,所述化合物iv的结构式为:所述第一溶剂由甲醇、乙醇、异丙醇、甘油、乙二醇、乙酸乙酯、二甲基四氢呋喃中的至少一种与叔丁醇的混合溶液。
[0024]
在一些实施方式中,所述第一溶剂中,所述叔丁醇的体积占第一溶剂体积的1/3~1/2;所述水和所述第一溶剂的体积比为1~3:1。
[0025]
在一些实施方式中,所述辅酶再生系统为葡萄糖和葡萄糖脱氢酶按一定比例的混合物,所述化合物iv、酮还原酶、酯水解酶、辅酶、葡萄糖和葡萄糖脱氢酶的质量比为1:0.1~0.3:0.2~0.4:0.005~0.015:0.1~0.3:0.1~0.3;所述化合物iv和所述水的质量比为1:10~15。
[0026]
在一些实施方式中,反应温度为28~32℃。
[0027]
在一些实施方式中,所述化合物iv由化合物i依次经酯化反应、脱氢反应、酰基化反应制备而成;所述化合物i的结构式为
[0028][0029]
在一些实施方式中,所述化合物iv的制备包括以下步骤:
[0030]
s1、将化合物i、dmap、dcm、三乙胺混合,在30~35℃下加入对苯磺酰氯并保温进行酯化反应,得到化合物ii;
[0031]
s2、将所述化合物ii、无水甲醇、pts、原乙酸三甲酯混合后,升温至30~35℃进行反应,得到化合物iii;
[0032]
s3、将所述化合物iii、醋酐、乙酰氯混合,升温至85~90℃进行反应,得到所述化合物iv;
[0033]
其中,所述化合物ii的结构式为所述化合物iii的结构式为:
[0034]
在一些实施方式中,该方法包括以下步骤:
[0035]
向所述第一反应容器中加入化合物iv、第一溶剂、水、酮还原酶、酯水解酶、辅酶、辅酶再生系统,控温至反应温度,使用na2co3调节ph=7.0~7.5,进行反应,检测无底物残留时停止反应,得到所述化合物v。
[0036]
在一些实施方式中,该方法还包括以下步骤:
[0037]
反应结束后,将反应体系升温至40~60℃,搅拌,再降温至15~25℃后过滤,用清水洗涤滤饼,在50~60℃下进行干燥,再加入无水乙醇并升温至50~60℃搅拌后趁热过滤,滤饼用乙醇淋洗,在30~45℃下负压浓缩,纯化水置换,浓缩干乙醇,水析出料,然后降温至10℃下进行析晶,过滤,干燥,得到化合物v。
[0038]
本发明还提供了一种7-脱氢胆固醇的制备方法,该方法包括上述任一项所述的中间体的制备步骤,具体为:以所述化合物iv为原料,通过上述任一项所述的方法制备得到化合物v,所述化合物v与格式试剂进行反应,得到7-脱氢胆固醇。
[0039]
在一些实施方式中,所述化合物v与所述格式试剂进行反应具体为:
[0040]
向第二反应容器中加入无水氯化铜、无水氯化里和无水四氢呋喃,搅拌溶清得第一试剂;
[0041]
向第三反应容器中加入镁屑、无水四氢呋喃,边搅拌边升温至60~70℃,加入卤代异戊烷,加入完毕后,保温进行反应至镁屑完全消失,得格式试剂;
[0042]
在-20~-10℃温度下,将所述第一试剂加入所述格式试剂中,然后加入所述化合物v,在-15~-10℃温度下进行反应,得到7-脱氢胆固醇。
[0043]
在一些实施方式中,所述卤代异戊烷包括溴代异戊烷、碘代异戊烷、氯代异戊烷中的一种。
[0044]
相较于现有技术,本发明的有益效果如下:
[0045]
本发明在7-脱氢胆固醇制备过程中,改变其中一个中间体的制备方法,以生物酶催化还原代替现有技术中的化学试剂还原,解决了现有技术中通过化学试剂进行还原过程中存在的异构体杂质多、收率低、对环境污染的问题,不仅提高了收率以及中间体产物的纯度,进一步提高了7-脱氢胆固醇的收率和纯度,而且对环境污染小,环保友好。本发明在制备该中间体的反应体系中,通过在反应体系中加入特定的极性溶剂,可以有效解决底物的三个共轭体系在酶催化反应过程中出现的结构不稳定的问题,在保证充分反应的同时提高目标中间体的收率,减少异构体杂质的生成,从而提高目标中间体的纯度,进而提高7-脱氢胆固醇的收率。
附图说明
[0046]
图1为实施例1中的化合物5的氢谱图;
[0047]
图2为实施例1中的化合物5的碳谱图;
[0048]
图3为实施例1中的含化合物5的白色晶体的液相色谱图;
[0049]
图4为实施例1得到的7-脱氢胆固醇产品的液相色谱图;
[0050]
图5为对比例1得到的含化合物5的白色晶体的液相色谱图。
具体实施方式
[0051]
在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
[0052]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
[0053]
本技术的下述实施例和对比例中,如无特别说明,所用试剂均为市购;其中,辅酶(nad+)为氧化型烟酰胺腺嘌呤二核苷酸,购自上海跃滕生物技术有限公司;所使用的酮还原酶为公开专利cn202110870850.7中的酮还原酶。
[0054]
实施例1
[0055]
一种7-脱氢胆固醇的制备方法,其工艺路线如下:
[0056][0057]
具体包括以下步骤:
[0058]
(1)化合物2的制备
[0059][0060]
室温下向反应瓶中加入100g中间体1、5gdmap(4-二甲氨基吡啶)、500ml dcm(二氯甲烷)以及100ml三乙胺,氮气置换,搅拌至溶清;升温至35℃,分5批向体系中加入对甲苯磺酰氯固体,每次间隔10-15min,每次加入15g,加完后保持35℃
±
2℃进行反应,tlc监控,约3h反应完毕;反应完毕后,将体系降至室温,加入300ml水,分液,有机相40℃减压浓缩,甲醇置换精制,浓缩至无dcm剩余,甲醇残留100ml左右,降温至0℃析晶1h,过滤,50℃干燥得141g化合物2,经检测,纯度大于98%。
[0061]
(2)化合物3的制备
[0062][0063]
室温下向反应瓶中加入100g中间体2、800ml无水甲醇、5gpts(对甲基苯磺酸)、80ml原乙酸三甲酯,升温至35℃
±
1℃进行反应,tlc监测,约3-4h反应完毕;反应完毕后,降温至-10~0℃搅拌1h,过滤,固体少量甲醇淋洗;接着固体用500ml的dcm、100ml水溶解,搅
拌下加入70g四氯苯醌,升温至40℃
±
1℃进行反应,tlc监测,约2h反应完毕;反应完毕后降至室温,过滤,固体用少量二氯甲烷淋洗;合并有机相,加入200ml亚硫酸钠溶液(内含30g亚硫酸钠)搅拌1h,分液,有机相40℃减压浓缩,甲醇置换精制,浓缩至无二氯甲烷残留,甲醇剩余100ml左右,降温至0℃析晶1h,过滤,固体50℃干燥,得93g中间体3,经检测,纯度大于94%。
[0064]
(3)化合物4的制备
[0065][0066]
室温下向反应瓶中加入100g中间体3、500ml醋酐,200ml乙酰氯,氮气置换,加热升温至85℃反应,tlc监测,约8h反应完毕,反应完后,减压浓缩至干,加入100ml甲醇淬灭剩余醋酐,而后40℃减压浓缩,甲醇置换成稠状,降温至0℃析晶1h,过滤,固体50℃干燥得95g淡黄色中间体4,经检测,纯度大于95%。
[0067]
(4)化合物5的制备
[0068][0069]
室温25℃,在洁净的500ml四口反应瓶中依次加入20.0g中间体4、40ml二甲基四氢呋喃、100ml叔丁醇、纯化水(140ml)、12g葡萄糖、12ml葡萄糖脱氢酶(菌液,400g/l)、12ml酮还原酶(菌液,400g/l)、20ml酯水解酶(菌液,400g/l)、0.2g辅酶nad+,控制温度30℃,使用10%的na2co3调ph=7.0,搅拌进行反应20小时,tlc检测无底物残留时停止反应(≤0.5%,tlc 254nm无荧光点);反应结束后将反应体系升温至40℃搅拌0.5小时,再降温至20℃,过滤,用20ml清水洗涤滤饼,于50℃热风循环烘箱中干燥,烘干后的滤饼重21.0g。滤饼加入100ml无水乙醇升温至50℃搅拌1小时后趁热过滤,滤饼用少量乙醇淋洗。将滤液在40℃,0.1mpa负压浓缩,纯化水置换,浓缩干乙醇、水析出料,将体系于10℃以下冷却,过滤,50℃热风循环烘箱中干燥,烘干后得白色晶体18.4g,重量收率92%,经检测(如图3所示),纯度>99%。其氢谱和碳谱图分别见图1和图2所示。
[0070]1h nmr(400mhz,cdcl3)δ7.78(d,2h),7.35(d,2h),5.56(m,1h),5.37(m,1h),3.96(m,1h),3.82(m,1h),3.63(m,1h),2.45(m,3h),2.27(m,1h),1.25-2.15(m,17h),0.98(m,6h),0.58(s,3h).
[0071]
13
c nmr(400mhz,cdcl3)δ144.64(s),140.55(s),140.10(s),133.14(s),129.79(s),127.93(s),119.49(s),116.65(s),75.53(s),70.39(s),54.06(s),51.50(s),46.11(s),43.02(s),40.77(s),38.85(s),38.34(s),37.01(s),36.53(s),31.97(s),27.34(s),
23.00(s),21.65(s),21.01(s),17.00(s),16.27(s),11.77(s).(两个碳信号峰与其他信号峰重叠)
[0072]
(5)7-脱氢胆固醇的制备
[0073][0074]
室温下向反应瓶中加入1g无水氯化铜、0.6g无水氯化锂和60ml无水thf(无水四氢呋喃),搅拌溶清得第一试剂;
[0075]
室温下向另一反应瓶中加入3.1g镁屑、100ml无水thf,搅拌下加热升温至60℃,缓慢滴加14.0g氯代异戊烷,滴加完保持60℃后继续反应直至镁屑消失(约2h),得格式试剂;
[0076]
将格式试剂降温至-20℃,向其中滴加第一试剂,全程保持体系温度低于-15℃,滴加完后-15℃保温反应30min,而后加入20g化合物5,使体系温度保持-10℃,保持该温度反应,tlc监测,约8h反应完毕;反应完毕后将反应液缓慢倒入200ml氯化铵水溶液中(含氯化铵15g),搅拌下加入200ml二氯甲烷提取,分液,40-50℃有机相减压浓缩,甲醇置换,置换至无dcm、thf残留,甲醇剩余20ml左右,降温至0℃析晶1h,过滤,固体少来了甲醇淋洗,50℃减压干燥得脱氢胆固醇18.1g,经检测(如图4所示),纯度大于98%。
[0077]
实施例2-6
[0078]
实施例2-6中,其他步骤相同,区别在于,化合物4进行制备化合物5的反应条件不同,具体反应条件和效果如表1所示:
[0079]
表1
[0080][0081][0082]
对比例1
[0083]
本对比例中,其他的制备方法与实施例1的相同,区别在于,化合物4制备化合物5的条件不同,其制备方法具体如下:
[0084]
室温25℃,在洁净的500ml四口反应瓶中依次加入20.0g中间体4、叔丁醇100ml、140ml纯化水、12ml葡萄糖、12ml葡萄糖脱氢酶(菌液,400g/l)、12ml酮还原酶(菌液,400g/
l)、20ml酯水解酶(菌液,400g/l)、0.2g辅酶nad+,控制温度30℃,使用10%的na2co3调ph=7.0;搅拌进行反应20小时,tlc检测无底物残留时停止反应(≤0.5%,tlc 254nm无荧光点);反应结束后将反应体系升温至40℃搅拌0.5小时,再降温至20℃,过滤,用20ml清水洗涤滤饼,于50℃热风循环烘箱中干燥,烘干后的滤饼重21.0g;滤饼加入100ml无水乙醇升温至50℃搅拌1小时后趁热过滤,滤饼用少量乙醇淋洗。将滤液在40℃,0.1mpa负压浓缩,浓缩到大量固体析出,将体系于0-5℃冷却析晶,过滤,50℃热风循环烘箱中干燥,烘干后得白色晶体17.6g,重量收率80%;经检测(如图5所示),纯度92.7%。
[0085]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0086]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1