一种匹莫范色林的制备方法与流程
1.本发明涉及化学制药技术领域,更具体地说,本发明涉及一种匹莫范色林的制备方法。
背景技术:
2.酒石酸匹莫范色林(哌马色林,pimavanserin tartrate),化学名为:n-[(4-氟苯基)甲基]-n-(1-甲基-4-哌啶基)-n
’‑
[[4-(2-甲基丙氧基)苯基]甲基]-脲,(2r,3r)-2,3-二羟基丁二酸盐(2:1),cas:706782-28-7;是美国阿卡迪亚(acadia)公司开发的一种新型非多巴胺神经递质类似物,主要用于治疗帕金森病患者伴发的错觉、幻觉、妄想等精神症状。
[0003]
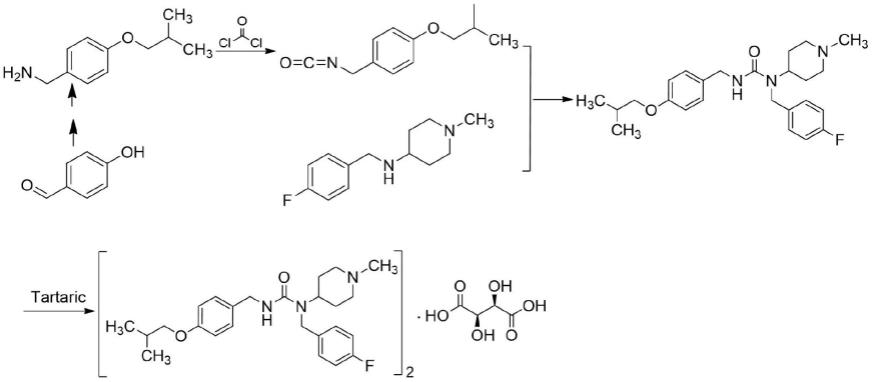
专利公开文献wo2006036874a1采用对羟基苯甲醛为起始物料,经醚化、肟化、还原合成4-异丁氧基苯甲胺,然后与光气进行酰化生成4-异丁氧基苄基异氰酸酯,再与n-(4-氟苄基)-1-甲基哌啶-4-胺进行脲化反应得到匹莫范色林,最后与酒石酸成盐反应制备得到酒石酸匹莫范色林,合成工艺路线如下:
[0004][0005]
上述工艺路线存在的缺陷为:合成路线较长,原子经济性不高;4-异丁氧基苯甲胺为液体原料不容易纯化,并且没有市售品;在制备4-异丁氧基苄基异氰酸酯时需要用到剧烈窒息性毒气——光气,增加了工业化生产的危险性和尾气吸收的设备投入,并且由于光气活性较高在制备得到4-异丁氧基苄基异氰酸酯的同时会生成对称脲杂质。
[0006][0007]
专利公开文献cn105153016a采用4-异丁氧基苯甲胺为原料,在甲苯中与羰基二咪唑(cdi)酰化得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺,然后再与n-(4-氟苄基)-1-甲基哌啶-4-胺缩合制备得到匹莫范色林,合成工艺路线
[0008][0009]
如下:
[0010]
上述工艺避免了剧毒原料——光气的使用,合成路线简短并且减少了环境污染。但是该路线也存在缺陷:4-异丁氧基苯甲胺没有市售品供应需要自行制备,该液体原料不稳定、不容易保存,很容易与空气中的二氧化碳反应生成碳酸盐;4-异丁氧基苯甲胺与羰基二咪唑(cdi)反应结束,未进行淬灭处理,残留的羰基二咪唑能继续参与反应从而产生其它杂质;并且n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺与n-(4-氟苄基)-1-甲基哌啶-4-胺缩合制备匹莫范色林过程中还会生成氧化杂质。
[0011][0012]
原料4-异丁氧基苯甲胺乙酸盐和原料n-(4-氟苄基)-1-甲基哌啶-4-胺市场供应充足、质量稳定,其中4-异丁氧基苯甲胺乙酸盐性状为固体粉末、市售品液相纯度为99.5%以上;n-(4-氟苄基)-1-甲基哌啶-4-胺市售品液相纯度为98.5%以上。两种原料能满足酒石酸匹莫范色林原料药大规模生产需要。
[0013]
文献《帕金森性精神病药物哌马色林的合成研究进展》(广东化工,2017,44(11):
177)中报道了采用市售易得的关键中间体4-异丁氧基苯甲胺乙酸盐与n-(4-氟苄基)-1-甲基哌啶-4-胺为起始原料制备酒石酸匹莫范色林的三条路线,其中方法2描述了采用4-异丁氧基苯甲胺乙酸盐为起始原料需要先碱化游离得到4-异丁氧基苯甲胺,然后与羰基二咪唑(cdi)反应得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺。现有碱化游离生产工艺采用4-异丁氧基苯甲胺乙酸盐在有机溶剂中加入氢氧化钠水溶液游离,反应结束分去水层,有机层水洗,回流分水脱除水分,降温后滴加入羰基二咪唑(cdi)的有机溶剂中进行反应,反应完全先后用水洗、饱和盐水洗、无水硫酸钠干燥、过滤滤液减压浓缩得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺。该制备工艺生产流程繁杂,经过碱化游离、水洗、干燥、浓缩等多步工序,会大幅增加制备生产周期和生产成本。
[0014][0015]
专利公开文献cn105820110a采用4-异丁氧基苯甲胺在n,n-二甲基甲酰胺和乙腈的混合溶剂中与羰基二咪唑(cdi)反应,反应结束后,蒸干溶剂,加入二氯甲烷萃取,干燥滤液减压浓缩得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺。该方法使用混合溶剂反应,采用两次浓缩,增加萃取工序,同样会增加原料成本及生产周期。
技术实现要素:
[0016]
鉴于此,本发明提供了一种匹莫范色林的制备方法,解决现有技术制备流程复杂、效率低、成本高、易引入杂质影响产品质量的缺陷。
[0017]
基于此,本发明的技术方案如下:
[0018]
一种匹莫范色林的制备方法,由4-异丁氧基苯甲胺乙酸盐与羰基二咪唑在碱液条件下酰化,再与n-(4-氟苄基)-1-甲基哌啶-4-胺在还原性溶剂下缩合反应,再经后处理得到。
[0019]
进一步地,所述匹莫范色林的制备方法,步骤包括:
[0020]
s1.4-异丁氧基苯甲胺乙酸盐和羰基二咪唑在非极性溶剂、有机碱作用下进行酰化反应,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的溶液;
[0021]
s2.向步骤s1产物溶液加入还原性溶剂、n-(4-氟苄基)-1-甲基哌啶-4-胺进行缩合反应,反应结束淬灭,依次用水洗、饱和盐水洗,再经浓缩、酯类溶剂置换,加入溶析剂,冷却析晶,过滤得到匹莫范色林。
[0022]
优选地,所述制备方法中,所述非极性溶剂选自二氯甲烷、甲苯或四氢呋喃。
[0023]
优选地,所述有机碱为有机胺。
[0024]
优选地,所述还原性溶剂为异丙醇。
[0025]
优选地,步骤s1中所述酰化反应完后加入纯化水洗涤,弃去水层。
[0026]
优选地,步骤s2中所述酯类溶剂为甲酸乙酯或乙酸乙酯。
[0027]
优选地,步骤s2中所述溶析剂为石油醚。
[0028]
本发明以上所述制备方法得到的匹莫范色林,hplc纯度大于99.5%,氧化杂质含量小于0.05%。
[0029]
本发明的有益效果在于:
[0030]
1.本发明所述制备方法采用市场供应充足、质量稳定的4-异丁氧基苯甲胺乙酸盐和n-(4-氟苄基)-1-甲基哌啶-4-胺为原料,省去现有技术采用n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺制备过程中碱化游离步骤(分层、水洗、回流脱水),以及酰化反应结束后采用简单的水洗分层替代现有技术中繁琐的水洗、饱和盐水洗、无水硫酸钠干燥、过滤、滤液减压浓缩等操作流程;向经过水洗分层得到的n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺溶液中加入还原性溶剂,可有效避免氧化杂质的生成。本发明方法操作简单,生产成本低,为工业化生产提供新的路径。
[0031]
2.本发明所述制备方法获得产物纯度大于99.5%,氧化杂质含量小于0.05%,质量稳定且可控,工业化生产可靠性强。
具体实施方式
[0032]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0033]
化学制药领域,药物合成路径不仅仅需要考虑产物的质量,如产物纯度、收率指标,也要考虑由于原材料选取导致中间过程杂质引入的风险及中间处理环节带来的影响。在工业生产中,应当充分考虑反应路径的整体性以及操作过程的便捷性及对于质量的影响,通常情况下仅仅通过常规已知的手段优化中间过程或是简单进行原料的替换是不能够保证的,另一方面基本化学反应原理虽为本领域人员熟知,但基于原料的选择对于整体性的影响存在不确定性,本领域技术人员在未掌握多种手段结合能实现提高质量的同时简化制备步骤的前提下,对于多种手段的尝试能否解决上述技术问题是不能预知的。因此,区别于现有技术的制备工艺,通过融合多种手段结合起来视为一个创造性的过程。
[0034]
针对现有技术采用4-异丁氧基苯甲胺(属苄胺类)存在原料不稳定、易与空气中的二氧化碳反应生成碳酸盐影响质量,以及缩合反应中易引入氧化杂质的缺陷,本发明通过原料、工艺多方面的优化得到合理解决。
[0035]
在一个实施例中,提出一种匹莫范色林的制备方法,由4-异丁氧基苯甲胺乙酸盐与羰基二咪唑在碱液条件下酰化,再与n-(4-氟苄基)-1-甲基哌啶-4-胺在还原性溶剂下缩合反应,再经后处理得到。具体步骤包括:
[0036]
s1.4-异丁氧基苯甲胺乙酸盐和羰基二咪唑在非极性溶剂、有机碱作用下进行酰化反应,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的溶液;
[0037]
s2.向步骤s1产物溶液加入还原性溶剂、n-(4-氟苄基)-1-甲基哌啶-4-胺进行缩合反应,反应结束淬灭,依次用水洗、饱和盐水洗,再经浓缩、酯类溶剂置换,加入溶析剂,冷
却析晶,过滤得到匹莫范色林。
[0038]
反应路线如下:
[0039][0040]
本方案选择4-异丁氧基苯甲胺乙酸盐作为原料,一方面市售质量可控,另一方面其在工业生产中不易与空气接触发生变质,但其选择可能会带来新的问题,4-异丁氧基苯甲胺乙酸盐在酰化反应过程前需脱去乙酸使得操作复杂化,中和采用的碱如果在酰化反应体系中加入,也需考虑可能对酰化反应的不利影响;另外缩合反应过程中由于反应强烈,极易氧化引入杂质。本方案选择有机碱液在溶剂体系中预先脱去乙酸,省去先碱化游离的操作,并且降低对酰化反应的影响;缩合反应在还原性溶剂下进行并且反应后进行了猝灭,有效降低氧化杂质的生成,所述制备方法得到的匹莫范色林,hplc纯度大于99.5%,氧化杂质含量小于0.05%。
[0041]
在一个优选的实施例中,所述非极性溶剂选自二氯甲烷、甲苯或四氢呋喃;有机碱为有机胺类,目的在于同加入的4-异丁氧基苯甲胺乙酸盐发生中和脱去乙酸,有机胺类优选烷基胺类,如三乙胺或二乙胺,以4-异丁氧基苯甲胺乙酸盐加入1g计,有机胺加入量为0.8~1.5ml。
[0042]
在一个优选的实施例中,所述还原性溶剂的目的是防止缩合反应过程中氧化杂质的引入,还原性溶剂优选异丙醇。
[0043]
在一个优选的实施例中,步骤s1中所述酰化反应完后加入纯化水洗涤,弃去水层,目的在于预先除去部分杂质。
[0044]
在一个优选的实施例中,步骤s2中所述酯类溶剂为甲酸乙酯或乙酸乙酯,所述溶析剂加入的目的为了析出产物,优选为石油醚。
[0045]
上述实施例中,所使用各类溶剂的用量为本领域人员可选择的使用范围,目的在于充分溶解形成反应或操作体系。
[0046]
以下为本发明较佳的实施例,用于验证发明的技术方案以及效果。
[0047]
实施例中,涉及的原料均为工业市售原料,标准为工业级,且相同原料纯度相同。
[0048]
实施例1
[0049]
向反应瓶内加入甲苯700ml、羰基二咪唑50g和三乙胺80ml,搅拌降温至5℃~10℃。加入4-异丁氧基苯甲胺乙酸盐60g并于5℃~10℃反应2小时。反应结束,有机层用纯化水100ml
×
3洗涤,弃去水层,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液;
[0050]
向上步反应得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液中加入异丙醇20ml,搅拌均匀后加入n-(4-氟苄基)-1-甲基哌啶-4-胺57.6g,升温至50℃~60℃保温反应4小时。反应结束,降温至20℃~30℃,加入纯化水100ml搅拌15分钟,静置分去水层,然后有机层依次用纯化水100ml
×
1洗涤、饱和盐水100ml
×
1洗涤,有机层减压浓缩去除溶剂,加入乙酸乙酯150ml升温至50℃~60℃溶解,滴加石油醚450ml,继续于50℃~60℃保温搅拌1
小时,降温至5℃~10℃析晶,析晶2小时,过滤、烘干得到匹莫范色林93.8g,hplc纯度99.93%,氧化杂质未检出。
[0051]
实施例2
[0052]
向反应瓶内加入二氯甲烷600ml、羰基二咪唑50g和二乙胺50ml,搅拌降温至5℃~10℃。加入4-异丁氧基苯甲胺乙酸盐60g并于5℃~10℃反应2小时。反应结束,有机层用纯化水100ml
×
3洗涤,弃去水层,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的二氯甲烷溶液;
[0053]
向上步反应得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的二氯甲烷溶液中加入异丙醇30ml,搅拌均匀后加入n-(4-氟苄基)-1-甲基哌啶-4-胺57.5g,于30℃~35℃保温反应8小时。反应结束,降温至20℃~25℃,加入纯化水100ml搅拌15分钟,静置分去水层,然后有机层依次用纯化水100ml
×
1洗涤、饱和盐水100ml
×
1洗涤,有机层减压浓缩去除溶剂,加入甲酸乙酯300ml升温至40℃~50℃溶解,滴加石油醚900ml,继续于40℃~50℃保温搅拌1小时,降温至5℃~10℃析晶,析晶2小时,过滤、烘干得到匹莫范色林91.9g,hplc纯度99.90%,氧化杂质的含量0.01%。
[0054]
实施例3
[0055]
向反应瓶内加入甲苯700ml、羰基二咪唑50g和二乙胺50ml,搅拌降温至5℃~10℃。加入4-异丁氧基苯甲胺乙酸盐60g并于5℃~10℃反应2小时。反应结束,有机层用纯化水100ml
×
3洗涤,弃去水层,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液;
[0056]
向上步反应得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液中加入异丙醇20ml,搅拌均匀后加入n-(4-氟苄基)-1-甲基哌啶-4-胺57.6g,升温至50℃~60℃保温反应4小时。反应结束,降温至20℃~30℃,加入纯化水100ml搅拌15分钟,静置分去水层,然后有机层依次用纯化水100ml
×
1洗涤、饱和盐水100ml
×
1洗涤,有机层减压浓缩去除溶剂,加入甲酸乙酯300ml升温至40℃~50℃溶解,滴加石油醚900ml,继续于40℃~50℃保温搅拌1小时,降温至5℃~10℃析晶,析晶2小时,过滤、烘干得到匹莫范色林93.3g,hplc纯度99.91%,氧化杂质未检出。
[0057]
对比例1
[0058]
将100g(0.558mol)4-异丁氧基苯甲胺加至500ml甲苯中,搅拌溶解,室温加入100g(0.627mol)羰基二咪唑,室温搅拌2小时,得到n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液。将124g(0.558mol)n-(4-氟苄基)-1-甲基哌啶-4-胺的500ml甲苯溶液滴加至上述制备得到的n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液中,升温回流5小时,反应结束后,降温至室温。
[0059]
反应体系中加入2.5l乙酸乙酯,用1l饱和食盐水洗涤两次,无水硫酸钠干燥。抽滤,滤液中加入10g活性碳,80-85℃搅拌脱色0.5小时,降温至室温,抽滤,40℃减压蒸除去溶剂,得到匹莫范色林粗品。取将100g匹莫范色林粗品加至500ml乙酸乙酯中,回流溶解,滴加1.0l正己烷,滴毕,降温至室温,室温搅拌4小时,抽滤,40~50℃减压干燥5小时,得淡黄色固体匹莫范色林。测得氧化杂质的含量0.17%。
[0060]
对比例2
[0061]
n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的制备方法及条件同实施例3,不同之处在于,向得到的n-(4-异丁氧基苯基)-1h-咪唑-1-甲酰胺的甲苯溶液中加入甲苯20ml,搅拌
均匀后加入n-(4-氟苄基)-1-甲基哌啶-4-胺57.6g,升温至50℃~60℃保温反应4小时。反应结束,降温至20℃~30℃,加入纯化水100ml搅拌15分钟,静置分去水层,然后有机层依次用纯化水100ml
×
1洗涤、饱和盐水100ml
×
1洗涤,有机层减压浓缩去除溶剂,加入甲酸乙酯300ml升温至40℃~50℃溶解,滴加石油醚900ml,继续于40℃~50℃保温搅拌1小时,降温至5℃~10℃析晶,析晶2小时,过滤、烘干得到匹莫范色林92.6g,hplc纯度99.52%,氧化杂质的含量0.15%。
[0062]
从实施例1-3的结果不难看出,本方案采用4-异丁氧基苯甲胺乙酸盐在有机胺条件下仍能够发生酰化反应,且省去了预先碱化游离的操作;缩合反应补加的还原性溶剂可有效抑制产生氧化杂质,由于该氧化杂质结构与产物匹莫范色林非常接近,因此在后处理过程中难以去除。对比例1作为现有技术,缩合过程中未加入还原性溶剂,导致最终产物存在氧化杂质,可能的原因是缩合反应过程中发生氧化反应,氧化杂质在后处理过程中难以分离,导致成品质量下降。对比例2作为实施例3的对比,更加显著的证明当还原性溶剂替换为甲苯溶剂时,保护作用消失,导致产生了氧化杂质。
[0063]
最后应说明的几点是,虽然上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明的基础上,以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1