一种CDK抑制剂的制备方法与流程
一种cdk抑制剂的制备方法
技术领域
1.本发明属于有机合成领域,具体涉及一种cdk抑制剂的制备方法。
背景技术:
2.细胞增殖是细胞分裂周期直接或间接失控的结果,细胞周期蛋白依赖性激酶(cdks)作为一种蛋白激酶,已知它们与各种细胞周期蛋白亚基相关,对细胞各种重要的调节通路起关键作用,包括细胞周期控制、细胞凋亡、神经元生理机能、分化和转录。
3.某些嘧啶类化合物已经被研究用于治疗包括癌症在内的细胞增殖性疾病和病症,例如4-噻唑-2-吡啶氨基-嘧啶和5-取代-4-噻唑-嘧啶(分别参见国际公开专利wo2005/012298和wo2013/156780)。这些化合物能抑制多种蛋白激酶,尤其是cdk。
4.本技术的发明人通过专利cn108349964a公开了一种新型的噻唑嘧啶化合物:n-环戊基-5-(2-((5-((4-乙基哌嗪-1-基)甲基)吡啶-2-基)氨基)-5-氟嘧啶-4-基)-4-甲基噻唑-2-胺化合物,实验表明,该化合物能够通过抑制cdk4和/或cdk6的活性来抑制细胞增殖,目前正处于临床研究阶段。
5.专利cn108349964a同时公开了n-环戊基-5-(2-((5-((4-乙基哌嗪-1-基)甲基)吡啶-2-基)氨基)-5-氟嘧啶-4-基)-4-甲基噻唑-2-胺化合物的制备方法,具体如下:
[0006][0007]
该制备方法中氟化反应的选择性差,反应中控显示中间体1约占55%,因剩余原料、杂质与产物性质类似,需经多次硅胶柱层析和重结晶才能得到纯度大于99.0%中间体1,该步反应总收率不到20%,且产生大量废液,环保压力较大;终产品的制备收率仅为29%,需微波照射操作,对设备的要求高,均不利于目标产物的工业化生产。
[0008]
因此,如何开发出一种高收率、高纯度且绿色环保的n-环戊基-5-(2-((5-((4-乙基哌嗪-1-基)甲基)吡啶-2-基)氨基)-5-氟嘧啶-4-基)-4-甲基噻唑-2-胺化合物的制备方法,是实现其工业化生产的关键所在。
技术实现要素:
[0009]
为了克服上述现有技术中的不足,本发明提供了一种cdk抑制剂n-环戊基-5-(2-((5-((4-乙基哌嗪-1-基)甲基)吡啶-2-基)氨基)-5-氟嘧啶-4-基)-4-甲基噻唑-2-胺的制备方法。
[0010]
本发明解决上述技术问题的技术方案如下:
[0011]
本发明提供了一种cdk抑制剂的制备方法,包括如下步骤:
[0012]
(1)在缚酸剂存在下,环戊基硫脲与1-氯丙酮反应生成式iia化合物;
[0013]
(2)式iia化合物经溴化剂取代生成式iib化合物;
[0014]
(3)在钯类催化剂和配体存在下,式iib化合物与双联频哪醇硼酸酯反应生成式iic化合物;
[0015]
(4)在钯类催化剂存在下,式iic化合物与2,4-二氯-5-氟嘧啶反应生成中间体ii;
[0016]
(5)在还原剂存在下,1-乙基哌嗪与2-硝基-5-醛基吡啶反应生成式iiia化合物;
[0017]
(6)式iiia化合物在氢气保护下经pd/c还原得到中间体iii;
[0018]
(7)在碱性试剂、钯类催化剂和配体存在下,中间体ii和中间体iii反应得到目标产物,即式i化合物;
[0019][0020]
进一步的,所述步骤(1)中缚酸剂选自吡啶、三乙胺或diea中的一种或几种,优选吡啶;所述环戊基硫脲与缚酸剂的摩尔比为1:1~1.5,优选1:1.1;
[0021]
进一步的,所述步骤(1)的反应温度为20~30℃,反应时间为3~6h;
[0022]
进一步的,所述步骤(2)中溴化剂选自br2、nbs或四溴化碳中的一种或几种,优选nbs;所述式iia化合物与溴化剂的摩尔比为1:1~1.2,优选1:1;
[0023]
进一步的,所述步骤(2)的反应温度为10~20℃,反应时间为1~2h;
[0024]
进一步的,所述步骤(3)中钯类催化剂选自醋酸钯、双(三苯基膦)氯化钯、三(二亚苄基丙酮)二钯中的一种或几种,优选醋酸钯;所述配体选自三环已基膦;所述钯类催化剂、配体、式iib化合物与双联频哪醇硼酸酯的摩尔比为0.05~0.15:0.1~0.2:1:1~2,优选0.1:0.18:1:1.5;
[0025]
进一步的,所述步骤(3)中还需加入醋酸钾;所述醋酸钾与式iib化合物的摩尔比
为2~4:1,优选3:1;
[0026]
进一步的,所述步骤(3)的反应温度为90~100℃,反应时间为5~8h;
[0027]
进一步的,所述步骤(4)中钯类催化剂选自醋酸钯、双(三苯基膦)氯化钯、三(二亚苄基丙酮)二钯中的一种或几种,优选双(三苯基膦)氯化钯;所述钯类催化剂与式iic化合物的摩尔比为0.05~0.15:1,优选0.1:1;
[0028]
进一步的,所述步骤(4)中还需加入碳酸钠,所述碳酸钠与式iic化合物的摩尔比为2~4:1,优选3:1;
[0029]
进一步的,所述步骤(4)的反应温度为溶剂回流温度,反应时间为2~6h;
[0030]
进一步的,所述步骤(5)中还原剂选自硼氢化钠、硼氢化钾、三乙基硼氢化钠或氢化锂铝中的一种或几种,优选三乙基硼氢化钠;所述还原剂与1-乙基哌嗪的摩尔比为1~1.1:1,优选1.05:1;
[0031]
进一步的,所述步骤(5)的反应温度为20~30℃,反应时间为8~12h;
[0032]
进一步的,所述步骤(6)中氢气微正压;所述反应温度为50~60℃,反应时间为5~8h;
[0033]
进一步的,所述步骤(7)中钯类催化剂选自醋酸钯、双(三苯基膦)氯化钯或三(二亚苄基丙酮)二钯中的一种或几种,优选三(二亚苄基丙酮)二钯;所述配体选自4,5-双二苯基膦-9,9-二甲基氧杂蒽;所述钯类催化剂、配体、中间体ii与中间体iii的摩尔比为0.03~0.08:0.1~0.2:1~1.2:1~1.2,优选0.05:0.15:1:1;
[0034]
进一步的,所述步骤(7)中碱性试剂选自碳酸钾、醋酸钠、碳酸铯或磷酸钾中的一种或几种,优选碳酸铯;所述碱性试剂、中间体ii与中间体iii的摩尔比为1~2:1~1.2:1~1.2,优选1.5:1:1;
[0035]
进一步的,所述步骤(7)的反应温度为95~100℃,反应时间为2~4h;
[0036]
进一步的,所述步骤(7)还包括将反应液趁热过滤、析晶、干燥步骤。
[0037]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0038]
本发明的有益效果在于:
[0039]
本发明通过全新的合成路线制备得到目标产物n-环戊基-5-(2-((5-((4-乙基哌嗪-1-基)甲基)吡啶-2-基)氨基)-5-氟嘧啶-4-基)-4-甲基噻唑-2-胺,同时开发出各关键中间体的合成路线。该方法反应条件温和、后处理方便,环保性好,对设备的要求低;筛选得到的反应条件(如催化剂、配体、碱性试剂)等提高了反应选择性,且有效保证了目标产物的收率和纯度,提高了产品质量,更有利于该cdk抑制剂的工业化生产。
附图说明
[0040]
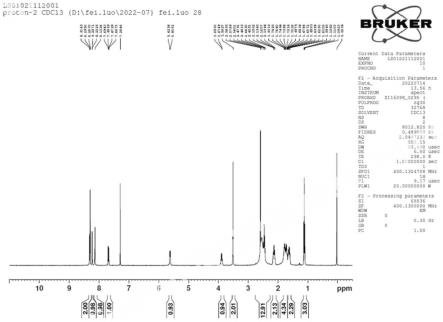
图1:实施例1制备的目标化合物氢谱图;
[0041]
图2:实施例1制备的目标化合物质谱图。
具体实施方式
[0042]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0043]
实施例1:
[0044]
式iia化合物的制备
[0045]
在10l反应釜中,分别称量144.2g环戊基硫脲(分子量144.24,1.0mol)、92.52g1-氯丙酮(分子量92.52,1.0mol)、87.0g吡啶(分子量79.10,1.1mol)及1442ml乙醇,搅拌,室温保温反应4h;将反应液倒入4326ml水中,室温搅拌1h;抽滤、滤饼干燥后得到155.3g所述式iia化合物(分子量182.29,0.852mol),纯度99.7%,摩尔收率85.2%。
[0046]
式iib化合物的制备
[0047]
在5l反应瓶中,分别称入182.3g式iia化合物(分子量182.29,1.0mol)及2735ml二氯甲烷,搅拌固体溶清;降温,10℃下分批加入178.0g nbs(分子量177.98,1.0mol),10~20℃保温1.5h;减压蒸除溶剂,加入2188ml 50%(v/v)乙醇/水溶液,室温搅拌1h;抽滤、滤饼干燥后得到227.5g所述式iib化合物(分子量261.18,0.871mol),纯度98.5%,摩尔收率87.1%。
[0048]
式iic化合物的制备
[0049]
在5l反应釜中,分别称入261.2g式iib化合物(分子量261.18,1.0mol)、380.9g双联频哪醇硼酸酯(分子量253.94,1.5mol)、294.4g醋酸钾(分子量98.14,3.0mol)、22.5g醋酸钯(分子量224.51,0.1mol)、50.5g三环已基膦(分子量280.43,0.18mol)及1946.4ml二甲亚砜,搅拌,氮气置换、保护;升温,90~100℃保温反应6h;趁热过滤,滤液室温析晶1h;抽滤、滤饼干燥后得到216.1g所述式iic化合物(分子量307.25,0.701mol),纯度98.2%,摩尔收率70.1%。
[0050]
中间体ii的制备
[0051]
在10l反应釜中,分别称入308.3g式iic化合物(分子量308.25,1.0mol)、167.0g2,4-二氯-5-氟嘧啶(分子量166.96,1.0mol)、318.0g碳酸钠(分子量105.99,3.0mol)、70.2g双(三苯基膦)氯化钯(分子量701.90,0.1mol)、1400ml水及4200ml dme,搅拌,升温至回流,保温4h;趁热过滤,滤液降至室温,保温析晶2h;抽滤、滤饼干燥后得到256.7g所述中间体ii化合物(分子量312.79,0.821mol),纯度98.9%,摩尔收率82.1%。
[0052]
式iiia化合物的制备
[0053]
在5l反应釜中,分别称入114.2g 1-乙基哌嗪(分子量114.19,1.0mol)、152.1g 2-硝基-5-醛基吡啶(分子量152.11,1.0mol)及3042ml二氯甲烷,搅拌,20℃下分批加入186.9g三乙基硼氢化钠(分子量177.98,1.05mol);后室温保温10h;滴加80ml水萃灭反应,减压蒸除溶剂,加入1710ml 75%(v/v)乙醇/水溶液,室温搅拌1h;抽滤、滤饼干燥后得到240.5g所述式iiia化合物(分子量250.30,0.961mol),纯度99.4%,摩尔收率96.1%。
[0054]
中间体iii的制备
[0055]
在10l反应釜中,分别称入250.3g式iiia化合物(分子量250.30,1.0mol)、25.0g钯碳及2003ml乙醇,搅拌,分别进行氮气置换、氢气置换、氢气微正压保压;升温、50~60℃保温6h;降温、室温滤除钯碳,体系加入4006ml水,室温搅拌1h;抽滤、滤饼干燥后得到216.6g所述中间体iii化合物(分子量220.32,0.983mol),纯度99.6%,摩尔收率98.3%。
[0056]
式i化合物(目标化合物)的制备
[0057]
在5l反应釜中,分别称入312.8g中间体ii(分子量312.79,1.0mol)、220.3g中间体iii(分子量220.32,1.0mol)、45.8g三(二亚苄基丙酮)二钯(分子量915.73,0.05mol)、
86.8g 4,5-双二苯基膦-9,9-二甲基氧杂蒽(分子量578.62,0.15mol)、448.73g碳酸铯(分子量325.82,1.5mol)及3128ml 1,4-二氧六环,搅拌,氮气置换、保护;升温,95~100℃保温反应3h;趁热过滤,滤液室温析晶1h;抽滤、滤饼干燥后得到454.9g所述目标化合物(分子量496.65,0.916mol),纯度98.6%,摩尔收率91.6%。
[0058]
通过核磁共振氢谱(1h-nmr)和质谱对上述制备的目标化合物检测,以验证其结构,见图1~2。其中所述结构表征包括但不限于hrms、h-nmr。
[0059]
核磁共振氢谱(1h-nmr)
[0060][0061]
表1目标化合物的氢谱解析数据
[0062][0063]1h-nmr(dmso-d6)给出15组峰积分比(由低场至高场)为1∶1∶1∶1∶1∶1∶1∶2∶3∶2∶8∶2∶4∶2∶3,共33个氢质子,与目标化合物的质子数相符。
[0064]
存在33个芳氢信号,分别为
⑴
δh 8.32(1h,s)与结构中11-nh-相符;
⑵
δh 8.30(1h,d)与结构中h
13
相符;
⑶
δh 8.22(1h,d)与结构中20-nh-相符;
⑷
δh 8.13(1h,s)与结构
中h
10
相符;(5)δh7.69(1h,dd)与结构中h7相符;
⑹
δh 5.62(1h,d)与结构中h8相符;
⑺
δh3.89(1h,m)与结构中h
21
相符;
⑻
δh 3.50(2h,s)与结构中h5相符;
⑼
δh 2.59(3h,d)与结构中h
18
相符;(10)δh 2.54(2h,q)与结构中h2相符;
⑾
δh 2.48(8h,br)与结构中2h3、2h4相符;
⑿
δh 2.15(2h,m)与结构中h
22
相符;
⒀
δh 1.80(4h,m)与结构中h
22
、h
23
相符;
⒁
δh 1.68(2h,m)与结构中h
23
相符;
⒂
δh1.13(3h,t)与结构中h1相符。
[0065]
质谱
[0066]
表2目标化合物的质谱数据
[0067][0068]
测试样品[m+h]
+
峰的质荷比为497,样品的分子量为496,与目标化合物的分子量相符;测试样品的分子量为偶数,分子中应含偶数个n原子,这与目标化合物含8个n原子相符。hrms测得样品的[m+h]
+
峰的测定质量为497.2719,与其理论值496.2533的误差为+18.6mda,其离子分子式为c
25h34
fn8s,故其分子式为c
25h33
fn8s,上述结果与目标化合物的分子式一致。
[0069]
综合核磁共振氢谱(1h-nmr)及hrms的数据解析,推定测试样品为目标化合物。
[0070]
实施例2:
[0071]
中间体ii的制备
[0072]
在10l反应釜中,分别称入308.3g式iic化合物(分子量308.25,1.0mol)、167.0g2,4-二氯-5-氟嘧啶(分子量166.96,1.0mol)、318.0g碳酸钠(分子量105.99,3.0mol)、22.5g醋酸钯(分子量224.51,0.1mol)、1400ml水及4200ml dme,搅拌,升温至回流,保温4h;趁热过滤,滤液降至室温,保温析晶2h;抽滤、滤饼干燥后得到189.4g所述中间体ii化合物(分子量312.79,0.606mol),纯度98.4%,摩尔收率60.6%。
[0073]
实施例3:
[0074]
式i化合物(目标化合物)的制备
[0075]
在5l反应釜中,分别称入312.8g中间体ii(分子量312.79,1.0mol)、220.3g中间体iii(分子量220.32,1.0mol)、45.8g三(二亚苄基丙酮)二钯(分子量915.73,0.05mol)、86.8g 4,5-双二苯基膦-9,9-二甲基氧杂蒽(分子量578.62,0.15mol)、144.17g叔丁醇钠(分子量96.11,1.5mol)及3128ml 1,4-二氧六环,搅拌,氮气置换、保护;升温,95~100℃保温反应3h;趁热过滤,滤液室温析晶1h;抽滤、滤饼干燥后得到398.2g所述目标化合物(分子量496.65,0.802mol),纯度91.6%,摩尔收率80.2%。
[0076]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1