一种1,2,3,4-四氢-9-甲基-4H-咔唑酮及其合成方法与流程
一种1,2,3,4-四氢-9-甲基-4h-咔唑酮及其合成方法
技术领域
1.本发明涉及化学合成领域,特别是一种1,2,3,4-四氢-9-甲基-4h-咔唑酮及其合成方法。
背景技术:
2.盐酸昂丹司琼其化学名为1,2,3,9-四氢甲基{(2-甲基咪唑-1-基)甲基}-4
‑ꢀ
氧代咔唑盐酸盐,其是一种选择性的5-羟色胺3(5-ht3)受体拮抗剂,其可通过拮抗外周迷走神经末梢和中枢化学感受区中的5-ht3受体,从而阻断因化疗和手术等因素促进小肠嗜铬细胞释放5-羟色胺,兴奋迷走传入神经而导致的呕吐反射。
3.目前,在工业化生产中四氢咔唑酮(1)或1,2,3,4-四氢-9-甲基-4h-咔唑酮(2)是制备盐酸昂丹司琼的关键原料,其产品质量直接决定最终产物的质量,但是,目前,四氢咔唑酮或1,2,3,4-四氢-9-甲基-4h-咔唑酮的合成工艺工艺粗放,产率低,合成产品质量差,限制了产品后续的使用范围。
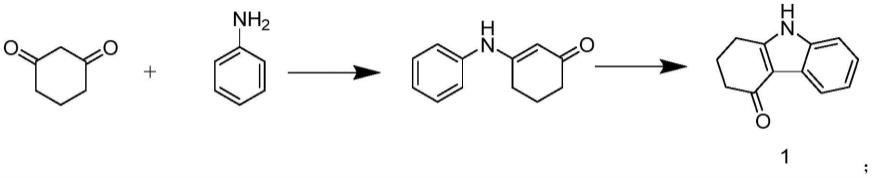
4.中国专利申请号cn201710375453.6公开了一种四氢咔唑酮的合成方法,涉及的合成路线如下式所示:
[0005][0006]
此合成路线以1,3-环己二酮和苯胺为原料,经过两步反应得到四氢咔唑酮,由四氢咔唑酮通过甲基化反应即得1,2,3,4-四氢-9-甲基-4h-咔唑酮,此反应用到的原辅料包括氧化铝、甲苯、醋酸钯等十余种,原辅料种类多,催化剂价格昂贵,且获得四氢咔唑酮还需要过硅胶柱,增加合成成本,影响产业化进程。
技术实现要素:
[0007]
本发明的目的是要解决现有技术中存在的1,2,3,4-四氢-9-甲基-4h-咔唑酮合成原辅料种类多,催化剂昂贵,得到目标产物需要过硅胶柱的问题,提供一种1,2,3,4-四氢-9-甲基-4h-咔唑酮及其合成方法。
[0008]
为达到上述目的,本发明是按照以下技术方案实施的:
[0009]
本发明的第一个目的是要提供一种1,2,3,4-四氢-9-甲基-4h-咔唑酮的合成方法,包括以下步骤:
[0010]
s1、取重量比为1∶1.6~2的2-环己烯-1-酮和四丁基三溴化铵,将2-环己烯-1-酮加入带有搅拌器、温度器的反应瓶中,分3~5次将四丁基三溴化铵加入到反应瓶中,加热至75~85℃,搅拌反应2~5h;
[0011]
s2、依次向步骤s1反应产物的反应瓶中加入2-环己烯-1-酮3~5倍重量的 2-卤
代-n-甲基苯胺、2-环己烯-1-酮1.1~1.7倍重量的碱及2-环己烯-1-酮0.002~ 0.012倍重量的催化剂,升温至110~120℃,搅拌反应3~4h,tlc跟踪反应,反应完冷却后过滤,滤液减压蒸馏,蒸出多余的2-卤代-n-甲基苯胺,剩余的固体用水洗涤至少3次,得到1,2,3,4-四氢-9-甲基-4h-咔唑酮。
[0012]
作为本发明的优选技术方案,所述碱为碳酸钾或碳酸钠。
[0013]
作为本发明的优选技术方案,所述2-卤代-n-甲基苯胺为2-氯-n-甲基苯胺或2-溴-n-甲基苯胺。
[0014]
作为本发明的优选技术方案,所述催化剂为氯化亚铜、溴化亚铜和碘化亚铜中的一种。
[0015]
本发明的第二个目的是要提供一种利用上述合成方法合成的1,2,3,4-四氢-9-甲基-4h-咔唑酮。
[0016]
与现有技术相比,本发明具有以下有益效果:
[0017]
1、本发明使用四丁基三溴化铵作为溴化试剂,加热后其可以直接溶于原料中参与反应,反应中不需要再另外加溶剂,节约环保高效,反应完的铵盐不用处理可以直接用于下一步的缚酸剂,大大节约了成本的同时,减少了三废的排放;
[0018]
2、本发明中2-溴(氯)-n-甲基苯胺的加入量是过量的,一是过量可以保证化合物3-溴-环己烷-1-酮能完全反应完;二是由于2-溴(氯)-n-甲基苯胺显碱性,可以作为缚酸剂,中和掉反应生成的hbr(hcl),有利于反应的充分进行;三是过量的2-溴(氯)-n-甲基苯胺可作为溶剂,避免了反应体系中使用其它溶剂,造成成本增加,带来二次污染;
[0019]
3、本发明的合成方法原料易得,操作方便,反应条件温和,制得的1,2, 3,4-四氢-9-甲基-4h-咔唑酮纯度高,收率高,收率可达93%以上,纯度高达99%以上,适合大规模生产。
附图说明
[0020]
图1是1,2,3,4-四氢-9-甲基-4h-咔唑酮的核磁氢谱图。
[0021]
图2是1,2,3,4-四氢-9-甲基-4h-咔唑酮的核磁碳谱图。
具体实施方式
[0022]
为使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步的详细说明。此处所描述的具体实施例仅用于解释本发明,并不用于限定发明。
[0023]
以下实施例中所用到的试剂及原料除特别说明外,均为市购,其具体来源不再详细记载。
[0024]
本发明的1,2,3,4-四氢-9-甲基-4h-咔唑酮合成路线为:
[0025]
[0026]
以下通过具体实施例详细阐述本发明的合成过程。
[0027]
实施例1
[0028]
将2-环己烯-1-酮1kg加入带有搅拌器、温度器的反应瓶中,分三批加入四丁基三溴化铵1.6kg,加热至75℃搅拌反应2h,然后依次往反应瓶中加入2-氯
ꢀ‑
n-甲基苯胺3kg、碳酸钾1.1kg及催化剂氯化亚铜0.002kg,升温温度110℃,搅拌反应3小时,tlc跟踪反应,反应完冷却后过滤,滤液减压蒸馏,蒸出多余的2-氯-n-甲基苯胺,剩余的固体用水洗涤3次,得到1,2,3,4-四氢-9-甲基-4h-咔唑酮1.83kg。为了确定制得的1,2,3,4-四氢-9-甲基-4h-咔唑酮的分子结构,对制得的1,2,3,4-四氢-9-甲基-4h-咔唑酮进行核磁共振检测,核磁氢谱图、核磁碳谱图分别如图1、图2所示,由图1、图2可知:m.p.192.7~194.1℃;1h nmr(300mhz,dmso-d6)δ:8.06~7.99(m,1h,-ar-h),7.61~7.46(m, 1h,-ar-h),7.38~7.09(m,2h,-ar-h),3.70(s,3h,-ch3),2.96(t,j=6.0 hz,2h,-ch
2-),2.39(t,j=5.8hz,2h,-ch
2-),2.17~2.01(m,2h,-ch
2-);
13
c nmr(75mhz,dmso-d6)δ:196.08,168.48,143.20,123.37,121.91,121.73, 119.78,119.56,110.97,39.12,30.57,22.29;hr(esi)ms(m/z):[m+h]
+
:200.0993。
[0029]
经计算,本实施例实际得到的1,2,3,4-四氢-9-甲基-4h-咔唑酮重量/理论上得到的化合物重量比即收率为88.3%,经高效液相色谱分析测得hplc纯度 97.5%。
[0030]
实施例2
[0031]
将2-环己烯-1-酮1kg加入带有搅拌器、温度器的反应瓶中,分三批加入四丁基三溴化铵1.8kg,加热至80℃搅拌反应3h,然后依次往反应瓶中加入2-溴
ꢀ‑
n-甲基苯胺4kg、碳酸钾1.5kg及催化剂溴化亚铜0.009kg,升温温度115℃,搅拌反应4小时,tlc跟踪反应,反应完冷却后过滤,滤液减压蒸馏,蒸出多余的2-溴-n-甲基苯胺,剩余的固体用水洗涤4次,得到1,2,3,4-四氢-9-甲基-4h-咔唑酮1.93kg,经计算,本实施例实际得到的1,2,3,4-四氢-9-甲基-4h
‑ꢀ
咔唑酮重量/理论上得到的化合物重量比即收率为93.2%,经高效液相色谱分析测得hplc纯度99.1%。
[0032]
实施例3
[0033]
将2-环己烯-1-酮1kg加入带有搅拌器、温度器的反应瓶中,分五批加入四丁基三溴化铵2.0kg,加热至85℃搅拌反应5h,然后依次往反应瓶中加入2-氯
ꢀ‑
n-甲基苯胺5kg、碳酸钠1.7kg及催化剂碘化亚铜0.012kg,升温温度120℃,搅拌反应3.5小时,tlc跟踪反应,反应完冷却后过滤,滤液减压蒸馏,蒸出多余的2-卤代-n-甲基苯胺,剩余的固体用水洗涤5次,得到1,2,3,4-四氢-9
‑ꢀ
甲基-4h-咔唑酮1.88kg,经计算,本实施例实际得到的1,2,3,4-四氢-9-甲基
ꢀ‑
4h-咔唑酮重量/理论上得到的化合物重量比即收率为90.7%,经高效液相色谱分析测得hplc纯度98.4%。
[0034]
实施例4
[0035]
将2-环己烯-1-酮1kg加入带有搅拌器、温度器的反应瓶中,分四批加入四丁基三溴化铵1.8kg,加热至80℃搅拌反应4h,然后依次往反应瓶中加入2-氯
ꢀ‑
n-甲基苯胺4kg、碳酸钠1.5kg及催化剂氯化亚铜0.009kg,升温温度110℃,搅拌反应3小时,tlc跟踪反应,反应完冷却后过滤,滤液减压蒸馏,蒸出多余的2-氯-n-甲基苯胺,剩余的固体用水洗涤6次,得到1,2,3,4-四氢-9-甲基-4h-咔唑酮1.85kg,经计算,本实施例实际得到的1,2,3,4-四氢-9-甲基-4h
‑ꢀ
咔唑酮重量/理论上得到的化合物重量比即收率为89.2%,经高效液相色谱分
析测得hplc纯度98.1%。
[0036]
本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1