一种L-肌肽的合成方法与流程
一种l-肌肽的合成方法
技术领域
1.本发明属于药物化学合成技术领域,具体涉及一种l-肌肽的合成方法。
背景技术:
2.l-肌肽(l-carnosine)是由β-丙氨酸和l-组氨酸缩合的一种天然的水溶性二肽,广泛地存在于脊椎动物的骨骼肌和中枢神经组织中,同时在心肌、肾、胃和嗅球等组织器官中也有存在,可简称β-丙氨酰-l-组氨酸,l-肌肽主要具有抗氧化功能,不仅能抑制金属离子和自由基的促氧化作用、供氢、供电子能力等活性,还能降低酯质氧化产物的浓度,及预防溃疡等重要的生理、药理功能,对于多种疾病如高血压、心脏病、老年痴呆、白内障、抗肿瘤等都具有一定的生物活性,在医药保健、食品及美容等领域具有非常广泛的应用。
3.有关l-肌肽化学合成方法,公开的文献及专利报道的比较多,归纳起来主要有两大类:(1)β-丙氨酸经氨基保护、羧基活化,再与保护的l-组氨酸缩合,最后脱保护基团得到l-肌肽。主要利用邻苯二甲酸酐与β-丙氨酸生成邻苯二甲酰-β-丙氨酸保护氨基,羧基与氯化亚砜反应生成邻苯二甲酰-β-丙氨酰氯,再与保护的l-组氨酸形成肽键、然后脱氨基的保护基团得到l-肌肽。该路线过程需要用到水合肼进行肼解邻苯二甲酰基脱保护,肼类属高毒类化合物,肼残留对产品质量影响大,而且肼解反应条件较苛刻,产生的副产物及杂质较复杂,使得收率较低,肽键形成过程中易消旋,影响产品纯度,溶剂消耗较大,不利于环保要求。
4.(2)使用不同的β-丙氨酸类似物或相似结构,构建与l-组氨酸缩合形成肽键,再进行官能团转化得到l-肌肽。常用的方法是在醇钠条件下,氰乙酸乙酯与l-组氨酸发生酰胺化反应,得到氰基乙酰-l-组氨酸,其氰基经催化氢化还原得到l-肌肽。该路线步骤较短,省去对不同基团的保护和脱保护的过程,避免消旋产生,但是醇钠条件需要无水操作,增加工业化难度,而且所用的氰乙酸乙酯为有毒物质,生产过程易造成环境污染。
5.针对现有工艺方法中存在的缺陷和不足,力图开发工艺简洁、经济环保的l-肌肽合成技术,尤其是寻求能够适应工业化生产的工艺方案,对该品种的经济和社会效益提高有着重要的现实意义。
技术实现要素:
6.针对现有技术的不足,本发明的目的在于提供一种l-肌肽的合成方法,该方法工艺条件温和,有助于降低制备成本,并且有益于体现高效绿色环保而得以满足工业化放大生产的要求。
7.为达此目的,本发明采用以下技术方案:一方面,本发明提供一种l-肌肽的合成方法,所述方法包括以下步骤:(1)3-氨基丙酸甲酯与2-(三甲基硅烷基)乙氧甲基氯反应,得到化合物int-1,反应式如下:
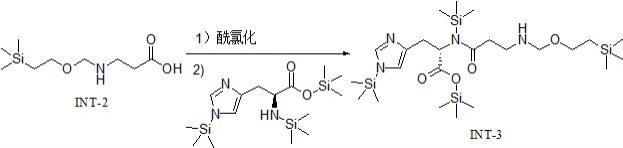
;(2)化合物int-1进行酯水解反应,得到化合物int-2,反应式如下:;(3)化合物int-2与酰氯化试剂进行酰氯化反应生成酰氯中间体,所述的酰氯中间体与n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯进行酰胺化反应,得到化合物int-3,反应式如下:;(4)化合物int-3进行脱保护反应,得到化合物int-4,反应式如下:;(5)在四丁基氟化铵存在下,化合物int-4进行脱保护反应,得到l-肌肽,反应式如下:。
8.本发明的合成路线条件温和,不使用毒性大的试剂,绿色环保,有利于终产品原料药的质量控制和提高,所用试剂原料易得,并且收率达90%,技术方案合理并且对环境友好,可以大量生产来满足使用需求,适用于工业化生产。
9.优选地,步骤(1)所述3-氨基丙酸甲酯与2-(三甲基硅烷基)乙氧甲基氯的摩尔比为1:1-2,例如1:1、1:1.2、1:1.4、1:1.5、1:1.7、1:1.9或1:2。
10.优选地,步骤(1)所述反应在碱性物质存在下进行。
11.优选地,所述碱性物质为氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、碳酸铯、氢化钠、三乙胺、n,n-二异丙基乙胺、吡啶、4-二甲氨基吡啶或2,6-二甲基吡啶中的任意一种或至少两种的组合。
12.优选地,所述碱性物质与3-氨基丙酸甲酯的摩尔比为1-4:1,例如1:1、1.5:1、1.8:1、2:1、2.5:1、2.8:1、3:1、3.5:1、3.8:1或4:1。
13.优选地,步骤(1)所述反应的溶剂为乙腈、四氢呋喃、甲基叔丁基醚、甲苯或n,n-二甲基甲酰胺中的任意一种或至少两种的组合。
14.优选地,步骤(1)所述反应的温度为20-50℃(例如20℃、25℃、30℃、35℃、40℃、45℃或50℃),反应时间为6-12小时(例如6小时、7小时、9小时、10小时、11小时或12小时)。
15.在本发明中,步骤(2)所述酯水解反应的方法和条件为本领域此类反应的常规方法和条件。
16.优选地,步骤(2)所述酯水解反应在碱性物质存在下进行,所述碱性物质为氢氧化钠、氢氧化钾或氢氧化锂中的任意一种或至少两种的组合。
17.优选地,步骤(2)所述酯水解反应的温度为30-100℃(例如30℃、40℃、50℃、60℃、70℃、80℃、90℃或100℃),时间为1-12小时(例如1小时、3小时、5小时、7小时、9小时或12小时)。
18.优选地,步骤(3)所述酰氯化试剂为草酰氯、氯化亚砜、氯甲酸异丁酯、三氯氧磷、五氯化磷、三氯化磷、硫酰氯、乙酰氯、氯乙酰氯、特戊酰氯或苯甲酰氯中的任意一种或至少两种的组合。
19.优选地,所述酰氯化试剂与化合物int-2的摩尔比为1-2:1,例如1:1、1.2:1、1.4:1、1.5:1、1.7:1、1.9:1或2:1。
20.优选地,步骤(3)所述酰氯化反应的温度为20-80℃(例如20℃、30℃、40℃、50℃、60℃、70℃或80℃),反应时间为1-6小时(例如1小时、2小时、3小时、4小时、5小时或6小时)。
21.优选地,步骤(3)所述化合物int-2与n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯的摩尔比为(1-1.2): (1-1.2),例如1:1、1:1.1、1:1.05、1:1.2、1.1:1或1.2:1。
22.优选地,步骤(3)所述酰胺化反应在碱性物质存在下进行。
23.优选地,所述碱性物质为n,n-二异丙基乙胺、三乙胺、二乙胺、乙二胺、吡啶、哌啶、三正丁胺、4-二甲氨基吡啶、2,6-二甲基吡啶、苯胺、苄胺、苯乙胺、n,n-二甲基苯胺、n,n-二乙基苯胺、三异丙胺、四甲基胍、二异丙胺、n-甲基吡咯烷酮、n-甲基吗啡啉、n-乙基吗啡啉、8-羟基喹啉、哌嗪、n-甲基哌嗪或二环己胺中的任意一种或至少两种的组合。
24.优选地,所述碱性物质与n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯的摩尔比为1-3:1,例如1:1、1.3:1、1.5:1、1.8:1、2:1、2.3:1、2.5:1、2.8:1或3:1。
25.优选地,步骤(3)所述酰胺化反应的溶剂为二氯甲烷、二氯乙烷、氯仿、四氢呋喃、甲基叔丁基醚、1,4-二氧六环或乙腈中的任意一种或至少两种的组合。
26.优选地,步骤(3)所述酰胺化反应的温度为50-80℃(例如50℃、55℃、60℃、65℃、70℃、75℃或80℃),反应时间为2-8小时(例如2小时、3小时、4小时、5小时、6小时、7小时或8小时)。
27.优选地,步骤(4)所述脱保护反应为水解脱保护反应或者醇解脱保护反应。
28.在本发明中,所述水解脱保护反应为加入水,控制温度为20-30℃(例如20℃、23℃、25℃、28℃或30℃),进行水解反应0.5-1小时(例如0.5小时、0.8小时或1小时)。
29.在本发明中,所述醇解脱保护反应是使用甲醇、乙醇或异丙醇中的任意一种或至少两种的组合进行。
30.在本发明中,所述醇解脱保护反应的温度为20-30℃(例如20℃、23℃、25℃、28℃或30℃),时间为0.5-1小时(例如0.5小时、0.8小时或1小时)。
31.在本发明中,所述醇解脱保护反应的溶剂为二氯甲烷。
32.优选地,步骤(5)所述四丁基氟化铵与化合物int-4的摩尔比为2-5:1,例如2:1、2.5:1、3:1、3.5:1、4:1、4.5:1或5:1。
33.优选地,步骤(5)所述脱保护反应的溶剂为四氢呋喃、甲基叔丁基醚、1,4-二氧六环或乙腈中的任意一种或至少两种的组合。
34.优选地,步骤(5)所述脱保护反应的温度为40-80℃(例如40℃、50℃、60℃、70℃或80℃),反应时间为2-6小时(例如2小时、3小时、4小时、5小时或6小时)。
35.作为优选技术方案,本发明所述的一种l-肌肽的合成方法包括以下步骤:(1)在碱性物质存在下,3-氨基丙酸甲酯与2-(三甲基硅烷基)乙氧甲基氯以摩尔比为1:1-2,于20-50℃,反应6-12小时,得到化合物int-1;(2)在碱性物质存在下,化合物int-1进行酯水解反应,得到化合物int-2;(3)化合物int-2与酰氯化试剂以摩尔比1:1-2在20-80℃进行酰氯化反应1-6小时,生成酰氯中间体,在碱性物质存在下,所述的酰氯中间体与n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯以摩尔比(1-1.2): (1-1.2)于50-80℃,进行酰胺化反应2-8小时,得到化合物int-3;(4)化合物int-3进行水解或醇解脱保护反应,得到化合物int-4;(5)在四丁基氟化铵存在下,化合物int-4在40-80℃进行脱保护反应2-6小时,所述四丁基氟化铵与化合物int-4的摩尔比为2-5:1,得到l-肌肽。
36.相对于现有技术,本发明具有以下有益效果:本发明的合成方法条件温和,不使用毒性大的试剂,绿色环保,有利于终产品原料药的质量控制和提高,所用试剂原料易得,并且收率高,技术方案合理并且对环境友好,可以大量生产来满足使用需求,适用于工业化生产。
具体实施方式
37.下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
38.实施例1在本实施例中,提供一种l-肌肽的合成方法,具体包括以下步骤:(1)制备化合物int-1:3-氨基丙酸甲酯(10g,97mmol)溶于乙腈(180ml),加入三乙胺(10g,99mmol),冰浴冷却,缓慢滴加2-(三甲基硅烷基)乙氧甲基氯(17g,0.1mol),升温20℃反应12h,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯-正庚烷混合溶剂重结晶,真空干燥,得到化合物int-1(20g),收率88%;1h nmr(dmso,400 mhz): (s,9h),0.80 (t,j=6.8 hz,2h),2.54 (t,j=7.1 hz,2h),2.91 (t,j=7.5 hz,2h),3.43 (t,j=7.1 hz,2h),3.69 (s,3h),4.81 (s,2h) ppm;(2)制备化合物int-2:化合物int-1(20g,86mmol)溶于乙醇(100ml)和水(70ml),冰浴冷却,缓慢加入氢
氧化钠(7g,0.18mol)的水(400ml)溶液,保温40℃反应1h,滴加10%醋酸溶液,加入二氯甲烷萃取,分层,收集有机相,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到化合物int-2(17g),收率90%;1h nmr(dmso,400 mhz):(s,9h),0.80 (t,j=6.8 hz,2h),2.52 (t,j=7.1 hz,2h),2.90 (t,j=7.5 hz,2h),3.42 (t,j=7.1 hz,2h),4.83 (s,2h) ppm;(3)制备化合物int-3:将化合物int-2(17g,77mmol)置于冰浴冷却下,滴加草酰氯(10g, 79mmol),搅拌下缓慢升温,至20℃反应6h,减压旋蒸浓缩至干得油状物,溶于二氯甲烷(500ml),加入n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯(371.70g,1mol),冰浴冷却,缓慢加入吡啶(7g,88mmol),升温80℃反应2h,减压旋蒸至干,二氯甲烷萃取,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到的粗品,经乙酸乙酯-石油醚混合溶剂重结晶,得到化合物int-3(38g),收率86%;1h nmr(dmso,400 mhz):(s,9h),0.08 (s,18h),0.20 (s,9h),0.80 (t,j=6.6 hz,2h),2.43 (t,j=7.5 hz,2h),2.90 (t,j=7.0 hz,2h),3.02 (m,2h),3.35 (t,j=6.9 hz,2h),4.57 (dd,2h),4.80 (m,1h),7.01 (s,1h),7.81 (s,1h) ppm。 (c=1.0,meoh);(4)制备化合物int-4:化合物int-3(38g,66mmol)溶于二氯甲烷(500ml),冰浴冷却,缓慢滴加异丙醇(80ml),保温20℃反应1h,抽滤,滤饼水洗,粗品经乙酸乙酯-石油醚混合溶剂重结晶,真空干燥,得到化合物int-4(22g),收率93%;1h nmr(dmso,400 mhz):(s,9h),0.80 (t,j=6.6 hz,2h),2.44 (t,j=7.5 hz,2h),2.91 (t,j=7.0 hz,2h),3.02 (m,2h),3.34 (t,j=6.9 hz,2h),4.58 (dd,2h),4.80 (m,1h),6.95 (s,1h),7.85 (s,1h) ppm。 (c=1.0,meoh);(5)制备l-肌肽:化合物int-4(22g,62mmol)溶于四氢呋喃(250ml),加入四丁基氟化铵(33g, 0.13mol),升温40℃反应6h,降至室温,抽滤过硅藻土,收集滤液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到l-肌肽(13g),收率93%;1h nmr(dmso,400 mhz): (t,j=7.5 hz,2h),3.03 (t,j=7.0 hz,2h),3.12 (m,2h),4.72 (m,1h),6.96 (s,1h),7.73 (s,1h) ppm。 (c=1.0,meoh)。
39.实施例2:(1)制备化合物int-1:3-氨基丙酸甲酯(15g,0.15mol)溶于四氢呋喃(200ml),加入碳酸钾(50g,0.36mol),冰浴冷却,缓慢滴加2-(三甲基硅烷基)乙氧甲基氯(36g,0.22mol),升温30℃反应9h,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯-正庚烷混合溶剂重结晶,真空干燥,得到化合物int-1(30g),收率88%;(2)制备化合物int-2:化合物int-1(30g,0.13mol)溶于甲醇(180ml)和水(90ml),冰浴冷却,缓慢加入氢氧化钾(15g,0.27mol)的水(100ml)溶液,保温30℃反应1h,滴加10%醋酸溶液,加入二氯甲烷萃取,分层,收集有机相,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到化合物int-2
(27g),收率96%;(3)制备化合物int-3:将化合物int-2(27g,0.12mol)置于冰浴冷却下,滴加氯化亚砜(22g,0.18mol),搅拌下缓慢升温,至50℃反应3h,减压旋蒸浓缩至干得油状物,溶于四氢呋喃(600ml),加入n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯(46g,0.12mol),冰浴冷却,缓慢加入三乙胺(25g,0.25mol),升温60℃反应5h,减压旋蒸至干,二氯甲烷萃取,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到的粗品,经乙酸乙酯-石油醚混合溶剂重结晶,得到化合物int-3(64g),收率91%;(4)制备化合物int-4:化合物int-3(60g,0.1mol)溶于二氯甲烷(800ml),冰浴冷却,缓慢滴加水(70g,3.9mol),保温20℃反应1h,抽滤,滤饼水洗,粗品经乙酸乙酯-石油醚混合溶剂重结晶,真空干燥,得到化合物int-4(36g),收率96%;(5)制备l-肌肽:化合物int-4(36g,0.1mol)溶于甲基叔丁基醚(500ml),加入四丁基氟化铵(90g,0.34mol),升温60℃反应4h,降至室温,抽滤过硅藻土,收集滤液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到l-肌肽(21g),收率92%。
40.实施例3:(1)制备化合物int-1:3-氨基丙酸甲酯(25g,0.24mol)溶于甲基叔丁基醚(400ml),加入氢氧化钾(54g,0.96mol),冰浴冷却,缓慢滴加2-(三甲基硅烷基)乙氧甲基氯(80g,0.48mol),升温50℃反应6h,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,粗品经乙酸乙酯-正庚烷混合溶剂重结晶,真空干燥,得到化合物int-1(52g),收率92%;(2)制备化合物int-2:化合物int-1(50g,0.21mol)溶于异丙醇(300ml)和水(150ml),冰浴冷却,缓慢加入氢氧化锂(10g,0.42mol)的水(200ml)溶液,保温25℃反应1h,滴加10%醋酸溶液,加入二氯甲烷萃取,分层,收集有机相,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到化合物int-2(45g),收率96%;(3)制备化合物int-3:将化合物int-2(40g,0.18mol)置于冰浴冷却下,滴加氯甲酸异丁酯(50g, 0.37mol),搅拌下缓慢升温,至80℃反应1h,减压旋蒸浓缩至干得油状物,溶于甲基叔丁基醚(600ml),加入n,1-双(三甲基硅基)-l-组氨酸三甲基硅基酯(68g,0.18mol),冰浴冷却,缓慢加入n,n-二异丙基乙胺(70g,0.54mol),升温50℃反应8h,减压旋蒸至干,二氯甲烷萃取,用食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到的粗品,经乙酸乙酯-石油醚混合溶剂重结晶,得到化合物int-3(91g),收率87%;(4)制备化合物int-4:化合物int-3(90g,0.16mol)溶于二氯甲烷(1000ml),冰浴冷却,缓慢滴加乙醇(200g,4.3mol),保温30℃反应0.5h,抽滤,滤饼水洗,粗品经乙酸乙酯-石油醚混合溶剂重结晶,真空干燥,得到化合物int-4(53g),收率95%;
(5)制备l-肌肽:化合物int-4(50g,0.14mol)溶于乙腈(600ml),加入四丁基氟化铵(180g,0.69mol),升温80℃反应2h,降至室温,抽滤过硅藻土,收集滤液,减压浓缩除去有机溶剂,加入二氯甲烷萃取,食盐水洗,无水硫酸钠干燥,减压旋蒸至干,得到l-肌肽(29g),收率91%。
41.申请人声明,本发明通过上述实施例来说明本发明的合成方法,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1