一种二羟甲基丁醛制备方法与流程
1.本技术涉及醛类化合物制备技术领域,具体而言,涉及一种二羟甲基丁醛制备方法。
背景技术:
2.2,2-二羟甲基丁醛(简称dmb)有两个羟甲基和一个醛基,化学性质活泼,dmb可以通过催化加氢制备三羟甲基丙烷(简称tmp),也可以通过催化氧化制备2,2-二羟甲基丁酸,是重要有机中间产物。但dmb不稳定,工业上很难分离提纯,所以通过dmb制备三羟甲基丙烷和2,2-二羟甲基丁酸的收率都不高。
3.dmb是由甲醛与正丁醛(简称nbd)在碱催化下发生羟醛缩合反应合成,此反应为可逆反应,由于一个正丁醛分子要与两个甲醛分子缩合,所以产物的收率并不是很高,目前工业上三羟甲基丙烷采用康尼查罗歧化法,加入过量的碱催化剂和过量的甲醛,产生的dmb后与甲醛发生歧化反应,生成三羟甲基丙烷和甲酸钠盐,由于缩合的产物dmb被转化为tmp使得反应继续向右进行,最后甲醛与nbd的反应转化率几乎可以达到95%以上。
4.为了解决这样的问题,提高dmb的转化率必须用有效的方法分离出产物dmb,使反应平衡能彻底进行。
5.而现有制备技术中,原料nbd的很大一部分反应产生不需要的副产物,dmb的收率低,不利于下游工序的合成,同时大量的副产物进入下游工序加氢或氧化后,会产生大量的没有经济价值的副产物,影响产品的分离,因此造成很大的经济负担。
技术实现要素:
6.本技术旨在至少解决现有技术中存在的技术问题之一。为此,本技术提出一种二羟甲基丁醛制备方法,该二羟甲基丁醛制备方法包括:
7.步骤一:将324克含量为37%的甲醛溶液加入1l的反应釜中启动搅拌,将144克正丁醛及59克含量为10%三甲胺溶液水溶液均匀滴加至反应釜中,控制滴加速度为2小时,滴加的过程中用水浴控制反应温度35~40℃,滴加结束后升温至40~45℃保温反应3小时;
8.步骤二:用甲酸中和反应液的ph至ph6.0-7.0;
9.步骤三:采用连续四级逆流萃取及四级逆流水洗实验槽,把得到的反应液用正丁醛及纯水进行萃取水洗,控制计量泵的进料流量,使萃取水洗体积比为:反应液-正丁醛-水为1:2:1;
10.步骤四:将得到的水洗油相经精馏塔精馏,采出部分正丁醛回用于萃取溶剂,当塔釜精馏温度上升至85℃时停止精馏。塔釜液返回至第一步缩合反应中代替正丁醛进行投料反应;
11.步骤五:萃取得到的水相经蒸发器减压蒸发,控制蒸发温度小于80℃,真空度8kpa,当萃取水相脱水至一半体积后停止蒸发取出蒸发水返回至萃取水洗代替纯水回用,蒸发控制蒸发真空度为3kpa,当蒸发釜内温度达到80℃时停止蒸发,取出浓缩液,即得到提
纯的dmb产用备用;
12.步骤六:多次重复步骤一到步骤五,分析数据并计算出dmb的反应转化率。
13.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤一中,甲醛溶液中的甲醛含量为4.0摩尔且相对于nbd为2.0摩尔,正丁醛为2.0摩尔,三甲胺为0.1摩尔且相对于nbd为0.05当量)。
14.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤一中,所用的有机碱催化剂为三甲胺、三乙胺及三丙胺。
15.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤一中,甲醛、正丁醛、催化剂的摩尔比为1.9~2.1:1:0.01~0.2。
16.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤三中,所用的萃取剂优选正丁醛。
17.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤三中,所用的萃取水洗体积比为:反应液-正丁醛-水为1:1~2.5:0.5~2。
18.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤4中,精馏塔精馏蒸发温度为50~80℃。
19.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤四中,精馏塔精馏蒸发真空度分两步,初步蒸发真空度为6~10kpa(绝压),第二步蒸发真空度为3~6kpa(绝压)。
20.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤三中,正丁醛萃取可有效地分离出未反应的原料及未反应完全的中间产物返回羟醛缩合反应。
21.根据本技术实施例的一种二羟甲基丁醛制备方法,在步骤三中,萃取及水洗程度至少四级以上,并且水洗得到的水洗油相中主要是分配比较大的甲醛、nbd、mmd、ecr等物质精馏出部分正丁醛后返回至步骤一进行缩合反应,萃取水相中主要是分配比较小的dmb及有机碱催化剂。
22.本技术的有益效果:
23.1、该方法采用了以羟醛缩合反应与萃取分离、蒸发分离等分离手段耦合的反应系统,将dmb转化效率提高百分之20-50之间,有效提高了反应的选择性。
24.2、该方法在dmb合成过程中产生的副产物mmd、ecr及原料nbd等与dmb在有机溶剂中的分配比不同,反应液可以通过溶剂萃取法进行分离合成的dmb,副产物mmd、ecr等,这些副产物再回至缩合系统进行反应。
25.总结:该方法根据美国公开专利us6171971记载,2,2-二羟甲基正丁醛的主反应历程如说明书附图3、4、5所示,并且制备过程中,中间产物mmb同时也会发生副反应,脱水形成2-乙基丙烯醛,止反应也是可逆的,并且而且正丁醛自身也会发生羟醛缩合反应形成2-乙基-3-羟基己醛,由以上可知正丁醛及甲醛的缩合副反应较多,只有把反应产物dmb分离出反应液才能有效提高反应形成dmb性;;
26.采用本发明方法所得到的二羟甲基丁醛具有高选择性和高收率,选择性为92~94%,收率为80%以上(基于正丁醛质量),而且反应温和,且因催化剂不溶于溶剂、活性组分无流失,所以无甲酸盐的产生,过简单分离即可得到高纯度的二羟甲基丁醛,可以用于直接加氢制备三羟甲基丙烷或氧化制备二羟甲基丁酸;
27.因此通过上述两个操作能够以一个新的反应系统在制备二羟甲基丁醛的同时,还
能够提高二羟甲基丁醛制备时dmb的转化效率,并且可以通过溶剂萃取法分离合成的dmb,以及其他副产物,将这些副产物缩合反应以后分离出反应液才能有效提高反应形成dmb增加了转化效率,dmb收益效率高,更有利于下游工序的合成,同时大量的副产物分离以后,没有经济价值的副产物会消失或转化为有经济效益的产物,有效地增加了二羟甲基丁醛制备后的产品、副产品的经济效益。
附图说明
28.为了更清楚地说明本技术实施例的技术方案,下面将对本技术实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本技术的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
29.图1是本发明中两种二羟甲基丁醛制备方法中gc分析结果数据含量对比的条形图;
30.图2本发明中两种二羟甲基丁醛制备方法中转化率数据含量对比的条形图;
31.图3是本发明中2,2-二羟甲基正丁醛的主反应过程第一部的反应方程式图;
32.图4是本发明中2,2-二羟甲基正丁醛的主反应过程第二部的反应方程式图;
33.图5是本发明中2,2-二羟甲基正丁醛的主反应过程第二部的反应方程式图;
34.图6是本发明实施例1中二羟甲基丁醛制备时dmb纯化含量的曲线图。
具体实施方式
35.下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行描述。
36.为使本技术实施方式的目的、技术方案和优点更加清楚,下面将结合本技术实施方式中的附图,对本技术实施方式中的技术方案进行清楚、完整地描述,显然,所描述的实施方式是本技术一部分实施方式,而不是全部的实施方式。基于本技术中的实施方式,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施方式,都属于本技术保护的范围。
37.下面参考附图描述根据本技术实施例的一种二羟甲基丁醛制备方法及制作方法,并且包括两个二羟甲基丁醛制备中计算dmb反应转化率的方法。
38.实施例1,
39.根据本技术实施例的一种二羟甲基丁醛制备方法,该方法包括以下步骤:
40.步骤一、将324克含量为37%的甲醛溶液(甲醛为4.0摩尔,相对于nbd为2.0当量)加入1l的反应釜中启动搅拌,将144克正丁醛(正丁醛为2.0摩尔)及59克含量为10%三甲胺溶液水溶液(三甲胺为0.1摩尔,相对于nbd为0.05当量)均匀滴加至反应釜中,控制滴加速度为2小时,滴加的过程中用水浴控制反应温度35~40℃,滴加结束后升温至40~45℃保温反应3小时。
41.步骤二、用甲酸中和反应液的ph至ph6.0-7.0
42.步骤三、采用连续四级逆流萃取及四级逆流水洗实验槽,把得到的反应液用正丁醛及纯水进行萃取水洗,控制计量泵的进料流量,使萃取水洗体积比为:反应液-正丁醛-水为1:2:1。
43.步骤四、得到的水洗油相经精馏塔精馏,采出部分正丁醛回用于萃取溶剂,当塔釜精馏温度上升至85℃时停止精馏。塔釜液返回至第一步缩合反应中代替正丁醛进行投料反应。
44.步骤五、萃取得到的水相经蒸发器减压蒸发,控制蒸发温度小于80℃,真空度8kpa,当萃取水相脱水至一半体积后停止蒸发取出蒸发水返回至萃取水洗代替纯水回用,第二步蒸发控制蒸发真空度为3kpa,当蒸发釜内温度达到80℃时停止蒸发,取出浓缩液,即得到提纯的dmb产用备用。
45.步骤六、按以上步骤重复多次计算出反应转化率。
46.实施例2,
47.根据本技术实施例的一种二羟甲基丁醛制备方法,该方法包括以下步骤:
48.步骤一、将308克含量为37%的甲醛溶液(甲醛为3.8摩尔,相对于nbd为2.0当量)加入1l的反应釜中启动搅拌,将144克正丁醛(正丁醛为2.0摩尔)及94克含量为10%三甲胺溶液水溶液(三甲胺为0.16摩尔,相对于nbd为0.08当量)均匀滴加至反应釜中,控制滴加速度为2小时,滴加的过程中用水浴控制反应温度35~40℃,滴加结束后升温至40~45℃保温反应3小时。
49.步骤二、用甲酸中和反应液的ph至ph6.0-7.0
50.步骤三、采用连续四级逆流萃取及四级逆流水洗实验槽,把得到的反应液用正丁醛及纯水进行萃取水洗,控制计量泵的进料流量,使萃取水洗体积比为:反应液-正丁醛-水为1:2.5:1.5。
51.步骤四、得到的水洗油相经精馏塔精馏,采出部分正丁醛回用于萃取溶剂,当塔釜精馏温度上升至85℃时停止精馏。塔釜液返回至第一步缩合反应中代替正丁醛进行投料反应。
52.步骤五、萃取得到的水相经蒸发器减压蒸发,控制蒸发温度小于80℃,真空度8kpa,当萃取水相脱水至一半体积后停止蒸发取出蒸发水返回至萃取水洗代替纯水回用,第二步蒸发控制蒸发真空度为3kpa,当蒸发釜内温度达到80℃时停止蒸发,取出浓缩液,即得到提纯的dmb产用备用。
53.步骤六、按以上步骤重复多次计算出反应转化率。
54.对于上述两个实施例中的补充,进一步的,在步骤一中,甲醛溶液中的甲醛含量为4.0摩尔且相对于nbd为2.0摩尔,正丁醛为2.0摩尔,三甲胺为0.1摩尔且相对于nbd为0.05当量)。
55.对于上述两个实施例中的补充,进一步的,在步骤一中,所用的有机碱催化剂为三甲胺、三乙胺及三丙胺。
56.对于上述两个实施例中的补充,进一步的,在步骤一中,甲醛、正丁醛、催化剂的摩尔比为1.9~2.1:1:0.01~0.2。
57.对于上述两个实施例中的补充,进一步的,在步骤三中,所用的萃取剂优选正丁醛。
58.对于上述两个实施例中的补充,进一步的,在步骤三中,所用的萃取水洗体积比为:反应液-正丁醛-水为1:1~2.5:0.5~2。
59.对于上述两个实施例中的补充,进一步的,在步骤4中,精馏塔精馏蒸发温度为50
~80℃。
60.对于上述两个实施例中的补充,进一步的,在步骤四中,精馏塔精馏蒸发真空度分两步,初步蒸发真空度为6~10kpa(绝压),第二步蒸发真空度为3~6kpa(绝压)。
61.对于上述两个实施例中的补充,进一步的,在步骤三中,正丁醛萃取可有效地分离出未反应的原料及未反应完全的中间产物返回羟醛缩合反应。
62.对于上述两个实施例中的补充,进一步的,在步骤三中,萃取及水洗程度至少四级以上,并且水洗得到的水洗油相中主要是分配比较大的甲醛、nbd、mmd、ecr等物质精馏出部分正丁醛后返回至步骤一进行缩合反应,萃取水相中主要是分配比较小的dmb及有机碱催化剂。
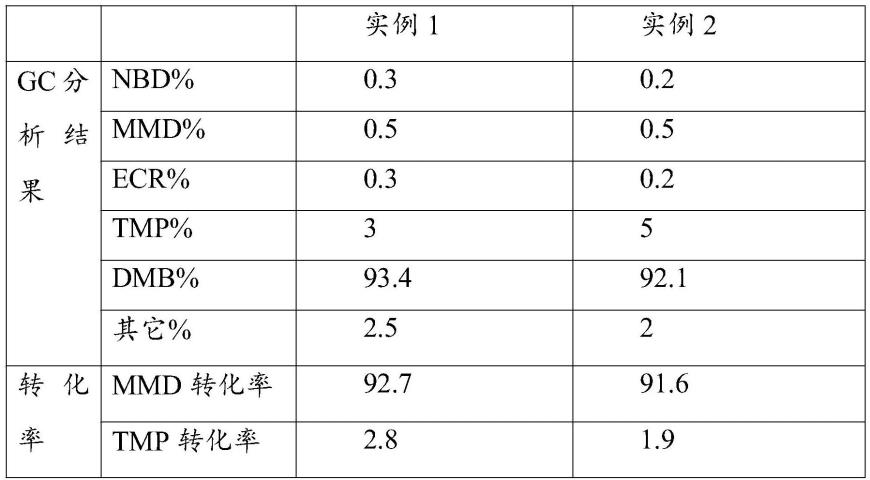
63.并且根据上述两个实施例的操作,得到的dmb浓缩液的gc分析的结果,以原料nbd为基准反应产生的tmp和dmb的收率计算结果见下表。
[0064][0065]
通过上述表格可以得知,两种改进的实施例方案中,dmb的转化率均高于百分之90以上,并且实施例1经过多次操作以后其dmb转化率数据更高,可以选择实施例1操作。
[0066]
并且表格中的重要组分nbd进行介绍:二环庚二烯,是一种有机化合物,化学式为c7h8,具有毒性,如遇到泄漏需要断火源,戴自给式呼吸器,穿一般消防防护服。在确保安全情况下堵漏。喷水雾可减少蒸发。用砂土或其它不燃性吸附剂混合吸收。然后运至空旷的地方掩埋、蒸发、或焚烧。如大量泄漏,利用围堤收容,然后收集、转移、回收或无害处理后废弃。
[0067]
该方法采用了以羟醛缩合反应与萃取分离、蒸发分离等分离手段耦合的反应系统,将dmb转化效率提高百分之20-50之间,有效提高了反应的选择性,并且该方法在dmb合成过程中产生的副产物mmd、ecr及原料nbd等与dmb在有机溶剂中的分配比不同,反应液可以通过溶剂萃取法进行分离合成的dmb,副产物mmd、ecr等,这些副产物再回至缩合系统进行反应
[0068]
2,2-二羟甲基正丁醛的主反应历程如说明书附图3、4、5所示,并且制备过程中,中间产物mmb同时也会发生副反应,脱水形成2-乙基丙烯醛,止反应也是可逆的,并且而且正丁醛自身也会发生羟醛缩合反应形成2-乙基-3-羟基己醛,由以上可知正丁醛及甲醛的缩
合副反应较多,只有把反应产物dmb分离出反应液才能有效提高反应形成dmb性;
[0069]
采用本发明方法所得到的二羟甲基丁醛具有高选择性和高收率,选择性为92~94%,收率为80%以上(基于正丁醛质量),而且反应温和,且因催化剂不溶于溶剂、活性组分无流失,所以无甲酸盐的产生,过简单分离即可得到高纯度的二羟甲基丁醛,可以用于直接加氢制备三羟甲基丙烷或氧化制备二羟甲基丁酸;
[0070]
因此通过上述两个操作能够以一个新的反应系统在制备二羟甲基丁醛的同时,还能够提高二羟甲基丁醛制备时dmb的转化效率,并且可以通过溶剂萃取法分离合成的dmb,以及其他副产物,将这些副产物缩合反应以后分离出反应液才能有效提高反应形成dmb增加了转化效率,dmb收益效率高,更有利于下游工序的合成,同时大量的副产物分离以后,没有经济价值的副产物会消失或转化为有经济效益的产物,有效地增加了二羟甲基丁醛制备后的产品、副产品的经济效益。
[0071]
以上仅为本技术的实施例而已,并不用于限制本技术的保护范围,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。应注意到:相似的标号和字母在下面的附图中表示类似项,因此,一旦某一项在一个附图中被定义,则在随后的附图中不需要对其进行进一步定义和解释。
[0072]
以上所述,仅为本技术的具体实施方式,但本技术的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本技术揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本技术的保护范围之内。因此,本技术的保护范围应所述以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1