一种泊马度胺衍生物其制备方法和应用
1.本发明属于抗肿瘤药物合成技术领域,具体涉及一种泊马度胺衍生物及其制备方法和应用。
背景技术:
2.泊马度胺为口服小分子衍生物,具有免疫调节、抗血管生成及抗增殖活性。主要通过靶向一种泛素e3连接酶cereblon(crbn),降解淋巴转录因子ikaros(ikzf1)和淋巴转录因子aiolos(ikzf3),同时调节肿瘤坏死因子α、白细胞介素6及血管内皮生长因子,增强对cd4+和cd8+t细胞的刺激。2020年5月15日,美国食品药品管理局批准泊马度胺用于治疗与获得性免疫缺陷综合征(aids)相关且对高效抗逆转录病毒疗法(haart)耐药的卡波西肉瘤(ks)患者,以及人类免疫缺陷病毒(hiv)阴性的ks患者。泊马度胺是20多年来获批治疗ks的首个口服新药。是沙利度胺衍生物中应用非常广泛的一个。沙利度胺(如图2)又名反应停,是曾经风靡欧美、非洲、日本又令人谈之色变的镇定药物,它具有中枢抑制作用,曾经广泛用于缓解妇女妊娠反应中的恶心呕吐症状。但是后来发现服用过沙利度胺的产妇诞下的胎儿手和脚都有不同程度的畸形,而且还有大量胎儿在出生前就因严重畸形而死亡。但是由于沙利度胺本身抗炎、免疫调节、抗血管新生——抗肿瘤作用,仍然有大量学者继续研究沙利度胺,期望得到一种具有沙利度胺优秀性能而又无致畸和神经毒性的药物,因此泊马度胺应运而生。与后者相比,泊马度胺苯环上连接的氨基使得自身化学性质更加稳定,且比沙利度胺具有更强的免疫调节作用。并且在临床应用方面,泊马度胺比沙利度胺的安全性更高,不良反应更少,它几乎没有致畸性和神经毒性,对多种血液病和实体恶性肿瘤都有作用。特别是泊马度胺可以作为protac分子中重要的e3泛素配体,在目前新药开发方面有着非常重要的作用,因此我们再次基础上进行改造,希望能够得到抗肿瘤活性比较好的新型化合物。河南科大实验室跟河南湾流生物科技有限公司合作,开发一种泊马度胺衍生物,河南湾流主要进行化合物的合成,河南科大实验室进行小分子设计和生物活性测试。
技术实现要素:
3.本发明解决的技术问题是提供了一种泊马度胺衍生物的制备方法及应用。
4.本发明为解决上述技术问题采用如下技术方案,一种泊马度胺衍生物的制备方法,其特征在于具体步骤为:
5.(1)、进一步优选,步骤(1)的具体过程为:将一定量的2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮和2-(2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-胺加入n,n-二甲基甲酰胺中,搅拌溶解后再加入一定量的n,n-二异丙基乙胺,在室温条件下搅拌至原料反应完全后,将反应液倒入水中,然后用二氯甲烷萃取多次,合并有机相,浓缩
后经硅胶柱层析分离得到
6.进一步优选,步骤(2)的具体过程为:将一定量的2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮和2-(2-(2-(2-氯乙氧基)乙氧基)乙氧基)乙-1-胺加入n,n-二甲基甲酰胺和乙醚混合液中,反应温度置于0℃搅拌一段时间后搅拌状态下加入n,n-二异丙基乙胺,继续在0℃条件下搅拌一段时间后加入乙醚和叠氮化钠,升至室温搅拌一段时间后将反应液倒入水中,然后用二氯甲烷萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到
[0007][0008]
进一步优选,步骤(2)的具体过程为:把一定量的4-溴苯胺和1,5-二溴-3-甲基戊烷和碳酸钾加入到n,n-二甲基甲酰胺中,加热至100℃搅拌反应一段时间后加入氰化亚铜和磺化钛氰钴,在氮气保护下,加热至100℃反应一段时间后冷却至室温,加入硅藻土进行搅拌过滤,并将滤液浓缩,经硅胶柱层析分离得到n-(4-苯甲腈)-4-甲基哌啶;所述的4-溴苯胺与1,5-二溴-3-甲基戊烷与碳酸钾与氰化亚铜的投料量摩尔比为1:1:1:1.5;所述的4-溴苯胺与磺化钛氰钴的投料量质量比为10:1。
[0009]
进一步优选,步骤(3)的具体过程为:将一定量的4-氨基异吲哚啉-1,3-二酮和2-溴-1,5-二戊酸甲酯加入n,n-二甲基甲酰胺中,搅拌溶解后再加入碳酸钾,在室温条件下搅拌一段时间升温至70℃,反应完全后将反应液倒入水中,然后用二氯甲烷萃取多次,合并有机相,浓缩后硅胶柱层析分离得到
[0010]
进一步优选,步骤(4)的具体过程为:将一定量的2-(2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-碘和加入n,n-二甲基甲酰胺中,搅拌溶解后再加入氢氧化钡和碘化钾和氢氧化钾,在室温条件下搅拌一段时间后将反应体系加热至100℃,真空排除反应釜内的空气,向反应釜内通入氨气,使反应釜压力达到0.5mpa,热至50℃,搅拌反应至原料反应完全,然后将反应液倒入水中,然后用二氯甲烷萃取多次,合并有机
相,浓缩后经硅胶柱层析分离得到
[0011]
进一步优选,步骤(5)的具体过程为:将和间氨基苯乙炔或对氨基苯乙炔加入到水、四氢呋喃和叔丁醇体积比1:1:1的混合溶剂中,再加入硫酸铜,抗坏血酸钠,氮气保护,加热至80℃,回流反应至原料反应完全,然后再加入间氟异氰酸苯酯,反应一段时间后停止反应,用硅藻土过滤,滤液真空浓缩,用二氯甲烷和甲醇(v/v=15:1)薄层层析,得到目标化合物。
[0012]
本发明的技术优势:本发明依据4-氯苯胺具有定向且具有相当距离的活性位置,通过一锅法进行了烷基化成环,然后再引入醛基,进一步转化为酰胺得到n-(4-苯甲酰胺)-4-甲基哌啶,一锅法反应有效的提高了产物的收率。
附图说明
[0013]
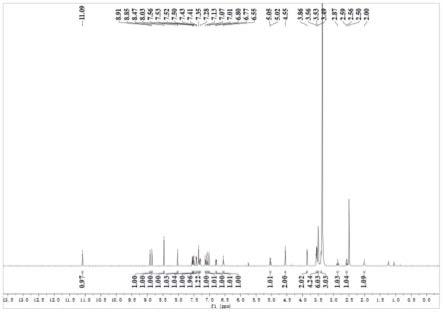
图1是实施例13制备得到的目标化合物的核磁氢谱图。
[0014]
图2是实施例14制备得到的目标化合物的核磁氢谱图。
[0015]
图3是实施例13化合物与brd4分子对接结果
具体实施方式
[0016]
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
[0017]
实施例1
[0018][0019]
在带有搅拌装置的反应瓶中,将2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮28g和2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-胺22g加入n,n-二甲基甲酰胺1000ml中,搅拌溶解后再加入n,n-二异丙基乙胺13g,在室温条件下搅拌120min后,将反应液倒入水1000ml中,搅拌30min后用二氯甲烷500ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(19.5g),ms(esi
+
)m/z:475[m+h]
+
。
[0020]
实施例2
[0021][0022]
在带有搅拌装置的反应瓶中,将2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮28g和2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-胺22g加入n,n-二甲基甲酰胺1000ml中,搅拌溶解后再加入n,n-二异丙基乙胺10g和n,n-二苯基甲酰胺2g,在室温条件下搅拌75min后,将反应液倒入水1000ml中,搅拌30min后用二氯甲烷500ml萃取多次,合并有机相,再用弱酸溶解调节有机相至中性,分出有机相,浓缩后经硅胶柱层析分离得到化合物2(34.7g),ms(esi
+
)m/z:475[m+h]
+
。
[0023]
实施例3
[0024][0025]
在带有搅拌装置的反应瓶中,将2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮28g和2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-胺22g加入n,n-二甲基甲酰胺1000ml中,搅拌溶解后再加入n,n-二异丙基乙胺20g,在室温条件下搅拌120min后,将反应液倒入水1000ml中,搅拌30min后用二氯甲烷500ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(22.9g),ms(esi
+
)m/z:475[m+h]
+
。
[0026]
实施例4
[0027][0028]
在带有搅拌装置的反应瓶中,将2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮28g和2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-胺22g加入n,n-二甲基甲酰胺1000ml中,搅拌溶解后再加入n,n-二异丙基乙胺26g,在室温条件下搅拌120min后,将反应液倒入水1000ml中,搅拌30min后用二氯甲烷500ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(28.17g),ms(esi
+
)m/z:475[m+h]
+
。
[0029]
实施例5
[0030][0031]
在带有搅拌装置的反应瓶中,将2-(2,6-二氧代-哌啶-3-基)-4-氟基-异吲哚-1,3-二酮28g和2-(2-(2-(2-氯乙氧基)乙氧基)乙氧基)乙-1-胺21.5g加入n,n-二甲基甲酰胺
800ml和乙醚200ml混合液中,反应温度置于0℃搅拌10min,然后搅拌状态下加入n,n-二异丙基乙胺26g,继续在0℃条件下搅拌30min,然后加入乙醚200ml和叠氮化钠20g,升至室温搅拌2h,将反应液倒入水1000ml中,然后用二氯甲烷500ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(32.91g),ms(esi
+
)m/z:475[m+h]
+
。
[0032]
实施例6
[0033][0034]
在带有搅拌装置的反应瓶中,把2-溴-1,5-戊二酸甲酯2.4g和4-氨基异吲哚啉-1,3-二酮1.7g(化合物3)加入n,n-二甲基甲酰胺150ml中,搅拌溶解后再加入碳酸钾1.4g,在室温条件下搅拌30min,升温至70℃,tlc监控原料反应完全,然后将反应液倒入水200ml中,然后用二氯甲烷50ml萃取多次,合并有机相,浓缩后硅胶柱层析分离得到化合物4(2.11g),ms(esi
+
)m/z:321[m+h]
+
。
[0035]
实施例7
[0036][0037]
在带有搅拌装置的反应瓶中,把2-溴-1,5-戊二酸甲酯2.4g和4-氨基异吲哚啉-1,3-二酮1.7g(化合物3)加入n,n-二甲基甲酰胺150ml中,搅拌溶解后再加入碳酸钾2.1g,在室温条件下搅拌30min,升温至100℃,tlc监控原料反应完全,然后将反应液倒入水200ml中,然后用二氯甲烷50ml萃取多次,合并有机相,浓缩后硅胶柱层析分离得到化合物4(2.35g),ms(esi
+
)m/z:321[m+h]
+
。
[0038]
实施例8
[0039][0040]
在带有搅拌装置的反应瓶中,把2-溴-1,5-戊二酸甲酯2.4g和4-氨基异吲哚啉-1,3-二酮1.7g(化合物3)加入n,n-二甲基甲酰胺150ml中,搅拌溶解后再加入碳酸钾2.8g,在室温条件下搅拌30min,升温至100℃,tlc监控原料反应完全,然后将反应液倒入水200ml中,然后用二氯甲烷50ml萃取多次,合并有机相,浓缩后硅胶柱层析分离得到化合物4(2.67g),ms(esi
+
)m/z:321[m+h]
+
。
[0041]
实施例9
[0042][0043]
在带有搅拌装置的高压反应釜中,将2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-碘3.3g和化合物4(3.2g)加入n,n-二甲基甲酰胺200ml中,搅拌溶解后再加入氢氧化钡3.5g和碘化钾1.7g和氢氧化钾0.56g,在室温条件下搅拌30min,然后将反应体系加热至100℃,真空排除反应釜内的空气,向反应釜内通入氨气,使反应釜压力达到0.5mpa,热至50℃,搅拌反应3h,tlc监控原料反应完全,然后将反应液倒入水200ml中,然后用二氯甲烷80ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(3.17g),ms(esi
+
)m/z:475[m+h]
+
。
[0044]
实施例10
[0045][0046]
在带有搅拌装置的高压反应釜中,将2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-碘3.3g和化合物4(3.2g)加入n,n-二甲基甲酰胺200ml中,搅拌溶解后再加入氢氧化钡3.5g和碘化钾1.7g和氢氧化钾0.56g,在室温条件下搅拌30min,然后将反应体系加热至100℃,真空排除反应釜内的空气,向反应釜内通入氨气,使反应釜压力达到0.3mpa,热至50℃,搅拌反应3h,tlc监控原料反应完全,然后将反应液倒入水200ml中,然后用二氯甲烷80ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(2.51g),ms(esi
+
)m/z:475[m+h]
+
。
[0047]
实施例11
[0048][0049]
在带有搅拌装置的高压反应釜中,将2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-碘3.3g和化合物4(3.2g)加入n,n-二甲基甲酰胺200ml中,搅拌溶解后再加入氢氧化钡3.5g和碘化钾1.7g和氢氧化钾0.56g和多孔沸石1g,在室温条件下搅拌30min,然后将反应体系加热至100℃,真空排除反应釜内的空气,向反应釜内通入氨气,使反应釜压力达到0.3mpa,热至50℃,保持压力不变,搅拌反应3h,tlc监控原料反应完全,然后将反应液倒入水200ml中,搅拌过滤后用二氯甲烷80ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(3.44g),ms(esi
+
)m/z:475[m+h]
+
。
[0050]
实施例12
[0051][0052]
在带有搅拌装置的高压反应釜中,将2-(2-(2-(2-叠氮基乙氧基)乙氧基)乙氧基)乙-1-碘3.3g和化合物4(3.2g)加入n,n-二甲基甲酰胺200ml中,搅拌溶解后再加入氢氧化钡3.5g和碘化钾1.7g和氢氧化钾0.56g和多孔氧化铝1g,在室温条件下搅拌30min,然后将反应体系加热至100℃,真空排除反应釜内的空气,向反应釜内通入氨气,使反应釜压力达到0.3mpa,热至50℃,保持压力不变,搅拌反应3h,tlc监控原料反应完全,然后将反应液倒入水200ml中,搅拌过滤后用二氯甲烷80ml萃取多次,合并有机相,浓缩后经硅胶柱层析分离得到化合物2(3.29g),ms(esi
+
)m/z:475[m+h]
+
。
[0053]
实施例13
[0054][0055]
在带有搅拌装置的反应瓶中,将化合物2(4.8g)和间氨基苯乙炔(1.2g)加入到水、四氢呋喃和叔丁醇体积比1:1:1的混合溶剂150ml中,再加入硫酸铜(1.6g),抗坏血酸钠(4.0g),氮气保护,加热至80℃,回流反应5h,tlc监控原料反应完全,然后再加入间氟异氰酸苯酯(1.4g)搅拌反应2h后停止反应,用硅藻土过滤,滤液真空浓缩,用二氯甲烷和甲醇(v/v=15:1)硅胶柱层析,得到目标化合物6.82g;1h nmr(600mhz,dmso-d6)δ11.09(s,1h),8.91(s,1h),8.85(s,1h),8.47(s,1h),8.03(s,1h),7.56-7.50(m,2h),7.43-7.41(m,1h),7.35-7.28(m,3h),7.13-7.07(m,2h),7.02(d,j=12.0hz,1h),6.80
–
6.77(m,1h),6.58
–
6.55(m,1h),5.05-5.02(m,1h),4.56
–
4.53(m,2h),3.86
–
3.84(m,2h),3.56
–
3.53(m,4h),3.50-3.48(m,6h),3.41-3.39(m,3h),2.90-2.84(m,1h),2.59-2.52(m,1h),2.03-2.00(m,1h).
[0056]
实施例14
[0057][0058]
在带有搅拌装置的反应瓶中,将化合物2(4.8g)和对氨基苯乙炔(1.2g)加入到水、四氢呋喃和叔丁醇体积比1:1:1的混合溶剂150ml中,再加入硫酸铜(1.6g,0.01mol),抗坏血酸钠(4.0g),氮气保护,加热至80℃,回流反应5h,tlc监控原料反应完全,然后再加入间氟异氰酸苯酯(1.4g)搅拌反应2h后停止反应,用硅藻土过滤,滤液真空浓缩,浓缩物用二氯
甲烷和甲醇(v/v=15:1)硅胶柱层析,得到目标化合物5.23g;1h nmr(600mhz,dmso)δ11.09(s,1h),8.93(s,1h),8.84(s,1h),8.41(s,1h),7.74(d,j=8.6hz,2h),7.57
–
7.48(m,4h),7.30(dd,j=15.1,8.2hz,1h),7.14
–
7.08(m,2h),7.02(d,j=7.0hz,1h),6.81
–
6.76(m,1h),6.58
–
6.55(m,1h),5.04(dd,j=12.8,5.5hz,1h),4.56
–
4.51(m,2h),3.86
–
3.82(m,2h),3.58
–
3.55(m,2h),3.53(d,j=5.6hz,2h),3.50(d,j=5.5hz,5h),3.41(dd,j=11.2,5.6hz,2h),2.91
–
2.83(m,1h),2.61
–
2.51(m,4h),2.04
–
1.98(m,1h).
[0059]
实施例15
[0060]
活性测试
[0061]
(1)细胞增殖抑制试验
[0062]
在苯基胺结构上,添加linker进行改造初步抑瘤实验结果。通过抗肿瘤活性实验,发现这类化合物抑制肿瘤细胞活性的效果普遍较好,实施例10得到的产物对食管癌细胞、肺癌细胞和多发性骨髓瘤细胞的ic
50
均在10μm以下。食管癌细胞的ic
50
值分别是,kyse30(8.83μm)、kyse150(12.71μm)、kyse450(4.45μm)。肺癌细胞的ic
50
值分别是,h460(22.66μm)、a549(19.51μm)。结肠癌细胞的ic
50
值分别是,hct116(10.64μm)、aog(5.55μm)。多发性骨髓瘤细胞的ic
50
值分别是,rpmi8226(1.89μm)、mm-1r(0.75μm)。实施例11得到的产物对食管癌细胞、肺癌细胞和多发性骨髓瘤细胞的ic
50
均在10μm以下。食管癌细胞的ic
50
值分别是,kyse30(8.49μm)、kyse450(7.31μm)。肺癌细胞的ic
50
值分别是,h460(10.59μm)、a549(》50μm)。多发性骨髓瘤细胞的ic
50
值分别是,rpmi8226(6.01μm)、mm-1r(5.45μm)。因此通过在在苯基胺结构上,添加linker进行改造是成功的。
[0063]
(2)细胞克隆形成实验
[0064]
平板克隆实验显示,实施例10得到的产物能明显抑制食管癌细胞kyse450和肺癌细胞h460、a549的增殖。泊马度胺抑制增殖效果不明显,该结果与mtt结果一致。
[0065]
(3)soft-agar克隆形成实验
[0066]
soft-agar克隆形成实验显示,实施例10得到的产物能明显抑制多发性骨髓瘤细胞rpmi8226、食管癌细胞kyse450和肺癌细胞h460的非依赖性增殖。泊马度胺抑制非依赖性增殖效果不明显,该结果与mtt结果和平板克隆结果一致。
[0067]
(4)细胞周期检测
[0068]
实施例10得到的产物能够阻滞多发性骨髓瘤细胞rpmi8226于g2/m期,阻滞食管癌细胞kyse450和肺癌细胞h460于g1期,而泊马度胺对肿瘤细胞的周期没有明显阻滞作用。
[0069]
(5)brd4和下游基因的蛋白表达变化情况
[0070]
随着作用时间和浓度的增加brd4的蛋白水平在癌细胞中呈下降趋势,其中kyse450细胞和h460细胞在24h,浓度为12μm时作用最明显。rpmi8226细胞在6h,浓度12μm时作用最明显。在蛋白水平检测c-myc在p4作用前后的变化情况。kyse450细胞和h460细胞在24h,浓度12μm时作用最明显。rpmi8226细胞在6h,浓度12μm时作用最明显。与brd4的趋势一致。相关性分析结果显示,brd4和c-myc的表达成显著正相关(r=0.94,p=0.015)。
[0071]
(6)动力学亲和力分析实验
[0072]
采用spr检测实施例10得到的产物与野生型brd4蛋白的结合。结果提示,实施例10得到的产物可与brd4野生蛋白结合,其kd(m)值为3.612e-5。说明实施例10得到的产物能够与野生型brd4蛋白结合。
[0073]
实施例16
[0074]
分子对接:实施例10化合物与brd4分子对接结果。图3的a-c:实施例10化合物与brd4的bd1结构域相互作用情况,autodock对接打分:-10.37;图3的d-f:实施例10化合物与brd4的bd2结构域相互作用情况,autodock对接打分:-7.41。实施例10化合物与brd4的bd1结构域的对接情况:实施例10化合物与pr082,lys91形成氢键;与trp81形成π-π共轭相互作用;与phe83,val87,leu92,ile146形成疏水相互作用(图3,a-c)。autodock对接打分:-10.37。实施例10化合物与brd4的bd2结构域的对接情况:实施例10化合物与pro375、his437形成氢键;与his437形成π-π共轭相互作用;与val380和val439形成疏水相互作用(图3,d-f)。autodock对接打分:-7.41。
[0075]
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1