一种多西他赛杂质及其制备方法与流程
1.本发明涉及药物合成制备技术领域,尤其是涉及一种多西他赛杂质及其制备方法。
背景技术:
2.多西他赛是半合成紫杉醇的衍生物,具有更广谱的抗白血病以及抗实体瘤活性,被认为是迄今为止疗效最显著的抗肿瘤药物之一。受到资源奇缺及全合成难度大的制约,商业化的紫杉醇类药物大多采用半合成方法。
3.为了合成纯度高的多西他赛,在多西他赛的质量研究中也需要对该杂质进行深入研究。暂未查得合成该杂质的相关文献报道。
技术实现要素:
4.鉴于高纯度多西他赛合成难度大,质量研究中对多西他赛杂质研究的需求。本发明针对多西他赛的研究,提供了一种多西他赛杂质的合成方法,本方法操作简单,反应条件温和,收率高,产品纯度好,所得产物对多西他赛质量研究有重要意义。
5.为了实现上述发明目的,本发明提供以下技术方案:
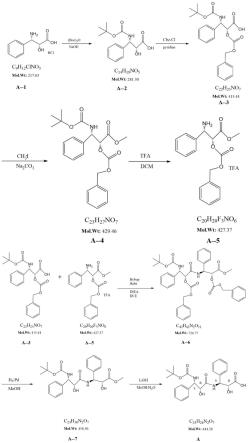
6.第一方面,本发明提供一种多西他赛杂质,如下图所示:
[0007][0008]
第二方面,本发明用于制备多西他赛杂质的底物如下图所示
[0009][0010]
本发明提供一种多西他赛杂质的制备方法,包括以下步骤:
[0011]
步骤1:取a-1所示化合物与碳酸酐二叔丁酯发生反应,浓缩,调节ph值,萃取,合并
有机相,洗涤,干燥,除去有机相,得白色固体产物,为a-2化合物;
[0012]
步骤2:取a-2化合物与氯甲酸苯甲基酯发生反应,浓缩,调节ph值,萃取后合并有机相,洗涤,干燥,浓缩,柱层析得无色油状物为a-3化合物;
[0013]
步骤3:取a-3化合物与碘甲烷发生sn2反应,萃取,合并有机相,洗涤,干燥,浓缩,柱层析除去溶剂得无色油状物为a-4化合物;
[0014]
步骤4:取a-4与三氟乙酸发生反应脱去保护基团,反应完全后浓缩,油泵抽干得白色片状固体为a-5所示化合物;
[0015]
步骤5:取a-3化合物和a-5化合物在缩合剂的作用下发生酰胺化反应,调节ph,萃取分液,洗涤,浓缩,柱层析得无色油状物为a-6化合物;
[0016]
步骤6:取a-6化合物在催化剂的作用下与氢气发生反应,催化氢解脱去保护基团,过滤,冲洗,浓缩,柱层析得白色固体为a-7化合物;
[0017]
步骤7:取a-7化合物与甲醇发生水解反应,反应完全后,调节ph值,浓缩,过滤,打浆,再次过滤,真空干燥得白色固体为a所示化合物。
[0018]
优选地,所述步骤1中,反应环境为碱性,碳酸酐二叔丁酯溶于四氢呋喃溶液中,a-1化合物与碳酸酐二叔丁酯的四氢呋喃溶液的质量比为1:(1-1.3)。
[0019]
优选地,步骤2中,氯甲酸苯甲基酯溶于四氢呋喃溶液中,a-2化合物与氯甲酸苯甲基酯的质量体积比为1g:(2.3-3)ml,柱层析的石油醚和乙酸乙酯的体积比为(10-1):1。
[0020]
优选地,步骤3中,a-3化合物与碘甲烷的质量比为1:(0.3-0.5),柱层析的乙酸乙酯和石油醚体积比为(50-5):1。
[0021]
优选地,步骤4中,a-4化合物与三氟乙酸的质量比为1:(3-5),反应溶剂为二氯甲烷。
[0022]
优选地,步骤5中,缩合剂为六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-羟基苯并三唑,a-3化合物与a-5化合物的质量比为1:(1-1.3),柱层析的乙酸乙酯和石油醚体积比为(15-5):1。
[0023]
进一步地,步骤5中,脱去的保护基团为步骤1中与碳酸酐二叔丁酯反应引入基团;
[0024]
优选地,步骤6中,催化剂为10%的钯炭,a-6化合物与甲醇的质量体积比为(40-50)g:1ml,柱层析二氯甲烷和甲醇的体积比为(50-20):1。
[0025]
优选地,步骤7中,a-7化合物与甲醇的质量体积比为(50-60)g:1ml。
[0026]
本发明的有益效果为:
[0027]
本发明提供一种多西他赛杂质的合成方法,该合成方法引入两种保护基团,在步骤4中脱去第一种保护基团生成的产物与步骤2产物反应,在步骤7脱去第二种保护基团生产目的产物,节省了原材料,缩短了反应周期,提高了产品纯度。该合成方法操作简单,反应条件温和,收率高,产品纯度好。
附图说明
[0028]
利用附图对本发明作进一步说明,但附图中的实施例不构成对本发明的任何限制,对于本领域的普通技术人员,在不付出创造性劳动的前提下,还可以根据以下附图获得其它的附图。
[0029]
图1为本发明所述多西他赛杂质的合成路线;
[0030]
图2为a-7化合物对应色谱图;
[0031]
图3为a化合物对应碳谱图;
[0032]
图4为a化合物对应氢谱图;
[0033]
图5为a化合物对应色谱图。
具体实施方式
[0034]
下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
[0035]
实施例1
[0036]
本发明的实施例涉及一种多西他赛杂质的制备,其合成方法包括以下步骤:
[0037]
step1:a-2的合成
[0038]
在250ml三口烧瓶中投入a-1化合物10g和100ml 1n naoh水溶液,搅拌降温至-5-0℃,缓慢滴加11.02g(boc)2o的thf溶液,然后缓慢滴加1n naoh水溶液保持体系的ph=8-9,直至tlc检测a-1化合物基本反应完全,然后缓慢滴加1n hcl水溶液调节ph≈7,浓缩掉thf,再用乙酸调节ph=3-4,用乙酸乙酯萃取三次,合并有机相,有机相用饱和nacl水溶液洗涤,用无水硫酸钠干燥,浓缩得12ga-2化合物,为白色固体(收率93.0%)。
[0039]
step2:a-3的合成
[0040]
在250ml三口烧瓶中投入a-2化合物12g和无水thf 120ml,氩气保护下搅拌降温至-5-0℃,滴加吡啶10.1g,滴加完毕后室温下搅拌半个小时。再在-5-0℃温度下缓慢滴加30ml cbz-cl的thf溶液,滴加完后,室温继续反应过夜。tlc检测基本反应完全,浓缩掉thf,往反旋蒸残留液中加入20ml 1n hcl稀盐酸调节ph=1-2,用乙酸乙酯萃取三次,合并有机相,有机相用饱和nacl水溶液洗涤,用无水硫酸钠干燥,浓缩得无色油状物,柱层析pe/ea=10/1-1/1得10.2g无色油状物a-3化合物(收率57.6%)。
[0041]
step3:a-4的合成
[0042]
在250ml三口烧瓶中投入无水dmf 100ml、a-3化合物6.2g和3.95g无水na2co3,氩气保护下,室温搅拌一个小时,然后降温至-5-0℃,缓慢滴加碘甲烷2.22g,滴加完毕,室温搅拌过夜。tlc检测a-3化合物反应完全,往反应液中加入水100ml,用乙酸乙酯萃取,合并有机相,有机相用饱和nacl水溶液洗涤四到五次,乙酸乙酯相再用无水na2so4固体干燥,浓缩去除溶剂得无色油状物,油状物柱层析(乙酸乙酯和石油醚=50/1-5/1),浓缩除去溶剂得5.1g无色油状物a-4化合物(收率79.7%)。
[0043]
step4:a-5的合成
[0044]
在100ml单口烧瓶中投入a-4化合物5.1g和二氯甲烷(溶剂)50ml,搅拌降温至-10-0℃,然后缓慢滴加三氟乙酸20g,滴加完毕后,室温继续反应3小时。tlc检测a-4化合物反应完全,浓缩掉dcm和三氟乙酸,油泵抽干,得5.1g白色片状固体为a-5化合物(收率100%),不用纯化直接用于下一步。
[0045]
step5:a-6的合成
[0046]
在250ml三口烧瓶中投入a-3化合物3.0g和dcm 30ml,氩气保护下,搅拌降温-5-0℃,然后分批加入7.5g的缩合剂pybop,再滴加3.7g a-5溶解在30ml二氯甲烷中的溶液以及
10-0℃,然后缓慢滴加三氟乙酸50.1g,滴加完毕后,室温继续反应3小时。tlc检测a-4化合物反应完全,浓缩掉dcm和三氟乙酸,油泵抽干,得10.2g白色片状固体为a-5化合物(收率100%),不用纯化直接用于下一步。
[0061]
step5:a-6的合成
[0062]
在250ml三口烧瓶中投入a-3化合物6.0g和dcm 60ml,氩气保护下,搅拌降温-5-0℃,然后分批加入15.0g的缩合剂pybop,再滴加7.8g a-5溶解在60ml二氯甲烷中的溶液以及8.5g的diea,搅拌10分钟后反应液变澄清,再分批加入4.0g的缩合hobt,所有物料加入完毕后,室温反应过夜。tlc检测a-3化合物大部分消失,往反应液中加入40ml 1n hcl水溶液调节ph=2-3,萃取分液,有机相再用饱和nahco3水溶液和饱和nacl水溶液依次洗涤,浓缩掉二氯甲烷得无色油状物,油状物经过两次柱层析(乙酸乙酯和石油醚=15/1-5/1),浓缩除去溶剂得3.2g无色油状物a-6化合物。
[0063]
step6:a-7的合成
[0064]
在100ml单口烧瓶中投入a-6化合物900mg和甲醇30ml,加入150mg(10%)钯炭,加上氢气球,置换三次然后室温反应过夜。tlc检测a-6化合物反应完全,反应液用硅藻土过滤,滤饼用30ml甲醇冲洗三次,浓缩掉甲醇得白色固体。固体柱层析(dcm/meoh=50/1-20/1)得500mg白色固体为a-7化合物。
[0065]
step7:a的合成
[0066]
在100ml三口烧瓶中投入a-7化合物500mg和甲醇8.3ml,搅拌降温-5-0℃,滴加60mg的lioh水溶液3ml,滴加完毕后,室温反应过夜,tlc检测反应完全。反应液用1n hcl稀盐酸调节ph=3-4,浓缩掉甲醇,析出大量白色固体,过滤,固体用30ml水打浆,过滤,真空干燥得425mg白色固体为a化合物。
[0067]
以上实施例对发明的实施方式作了详细说明,但发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1