pEcCas9-2.0质粒及其构建方法和应用
peccas9-2.0质粒及其构建方法和应用
技术领域
1.本发明涉及基因工程技术领域,具体涉及peccas9-2.0质粒及其构建方法和应用。
背景技术:
2.crispr-cas是许多细菌和古细菌中的获得性免疫系统,用来抵御外源遗传物质的入侵,目前已被开发成基因编辑工具并广泛用于各种生物中。cas9等cas家族蛋白质能在向导rna引导下切割宿主基因组导致宿主dna双链断裂,宿主主要通过同源重组和非同源末端连接两条途径对断裂的dna双链进行修复,然而大部分细菌缺乏内源的非同源末端连接途径。因此目前细菌中基于crispr-cas的基因敲除或插入工具多是通过外源提供修复模板,借助同源重组修复来实现,而在无外源修复模板情况下,crispr-cas引起的dna双链断裂通常是无法被修复的,并将导致细菌死亡。借助该原理,crispr-cas可被开发成抗菌剂用于清除微生物群落中特定菌株,在抗生素耐药性和精确微生物组工程的时代,crispr-cas抗菌剂有望对我们未来治疗疾病的方式产生重大影响。相对于抗生素而言,crispr-cas系统具有高度的特异性与方便的可编程性,能从菌株水平进行识别和消除目标菌株,而不影响原本的微生物群落。目前已有研究报道利用噬菌体来“输送”crispr-cas元件,以实现对肠道微生物中病原细菌如耐药性大肠杆菌进行清除;也有研究将其用于清除用于诊断和治疗各种疾病的工程益生菌,以防止工程益生菌在环境中的传播。
3.然而,在实际应用情况下,多数细菌在无外源修复模板情况下也能存活(即为逃逸子),比例约为10-6
至10-1
。美国国立卫生研究院强调:108个细胞中逃逸1个及以下的细胞被认为是可接受的清除效率(即逃逸率不能高于10-8
)。逃逸子的存在,不仅阻碍了crispr在抗菌剂方面的应用,同时也使得在细菌中进行基因编辑如基因敲除时的效率降低。因此了解逃逸的原因可加快高效抑菌剂的开发,也有助于高效基因编辑工具的开发。
4.在关于逃逸的机制研究方面,研究人员们主要从改造菌株和改造工具两个角度对逃逸进行了研究。
5.在改造菌株方面,有研究表明:大肠杆菌可以通过reca蛋白介导的sos反应对断裂dna进行修复上调易错dna聚合酶的表达,进而容易造成靶标dna的突变,导致使修复的新序列无法被原grna识别并进行二次切割,从而逃避了无修复同源臂下crispr-cas诱导的细胞死亡。易错dna聚合酶表达的上调在提高靶标dna突变率的同时,也会提高cas9蛋白等crispr相关基因的突变率,从而进一部提高细菌在cas9蛋白的切割中的逃逸率。因此,crispr-cas系统在reca突变体中的表达更有效。
6.但是基于crispr的抑菌剂或实现高效编辑的工具需从优化基因编辑工具本身入手,而非通过改造目标菌株,如敲除基因等来辅助工具以实现高效编辑。受限于逃逸机制的研究,目前仅有提高cas蛋白活性,筛选grna文库以确定染色体不同切割位置对逃逸率的影响以及抑制dna修复途径几种方法,例如在质粒中表达能与断裂dna双链两端结合的gam蛋白的基因,来抑制细菌对断裂基因组的修复进而降低逃逸率。由于dna双链断裂诱导的sos应答能够促进易错dna聚合酶导致的突变,使得细胞逃逸crispr-cas9靶向切割的概率提
高。有研究基于cas9与reca56共表达的质粒系统,将reca的突变体reca56叠加到cas9表达质粒上,通过reca56的引入,削弱了野生型reca的活性,进而不能正常开启易错dna聚合酶的表达的上调途径以此达到提高crispr-cas9介导的切割效率,减少逃逸的目的,但优化后工具的逃逸率仍高于10-8
。
技术实现要素:
7.为解决上述技术问题,本发明提供了peccas9-2.0质粒及其构建方法和应用,所述peccas9-2.0质粒在基因编辑中能够达到低于10-8
的逃逸率,在大肠杆菌中能够达到100%的基因编辑率。
8.本发明的第一个目的提供peccas9-2.0质粒,所述peccas9-2.0质粒包括表达cas9蛋白的基因cas9,λ-red重组系统,阿拉伯糖操纵子arac,p15a复制子,氯霉素抗性基因cmr。
9.本发明的第二个目的是提供所述peccas9-2.0质粒的构建方法,通过增加cas9基因的拷贝构建获得peccas9-2.0质粒,具体包括以下步骤:
10.cas9基因及其启动子与线性化的p15a进行组装(组装方法参照南京诺维赞公司的clonexpress multis one step cloning kit试剂盒),获得质粒p15a-cas9,然后再在p15a-cas9的基础上叠加λ-red重组系统相关基因,获得peccas-2.0质粒。
11.进一步地,所述的cas9基因的扩增引物包括p15a-cas9-up和p15a-cas9-dn;
12.所述p15a-cas9-up的核苷酸序列如seq id no.1所示;
13.所述p15a-cas9-dn的核苷酸序列如seq id no.2所示。
14.本发明的第三个目的是提供peccas9-2.0质粒在基因编辑中的应用。
15.进一步地,所述peccas9-2.0质粒能作为微生物中的基因组编辑工具。
16.进一步地,所述的peccas9-2.0质粒能够达到低于10-8
的逃逸率。
17.进一步地,所述的peccas9-2.0质粒在大肠杆菌mg1655中cada、maeb和gntt基因中的基因编辑率为100%。
18.本发明的第四个目的是提供peccas9-2.0质粒在制备抗菌剂中的应用。
19.与现有技术相比,本发明的有益效果在于:
20.1、本发明构建得到的peccas9-2.0质粒,在大多数位点都能够达到低于10-8
的逃逸率,在大肠杆菌mg1655的cada、maeb和gntt基因中能够达到100%的编辑率,在大肠杆菌的其他菌株中也同样表现良好,在大肠杆菌bl21(de3)菌株以及大肠杆菌w菌株的cada、maeb基因中的编辑效率都达到了100%,peccas9-2.0质粒促进crispr系统在抗菌剂开发中的应用,并可为研究其他细菌逃逸crispr-cas的编辑提供参考。
21.2、本发明通过检测大肠杆菌mg1655逃逸子crispr元件dna突变情况(包括cas9和grna),获得大肠杆菌逃逸cas9编辑的规律并揭示了大肠杆菌mg1655逃逸cas9编辑的主要原因之一,大部分质粒的cas9基因都有突变现象,除了点突变及个别单核苷酸的缺失和增添以外,还有is序列的插入;is序列是细菌基因组中的一类特殊序列,能够插入到其他基因序列中并使这部分基因失活,是逃逸出现的重要原因之一。
22.3、本发明以dna突变为研究重点,通过增加cas9基因的拷贝数,最终构建新质粒peccas9-2.0,质粒peccas9-2.0将使用具有更高拷贝数的p15a复制子,在不会对宿主大肠杆菌的生长造成影响的前提下拥有更多的拷贝数,能够在细菌体内表达更多的cas9蛋白,
rna,grna):在crispr-cas系统中起引导功能的rna片段,和cas蛋白结合为复合体,引导cas蛋白切割目标序列;(4)sos反应:sos反应是在大肠杆菌中发现的第一个dna修复系统,它是在细菌dna遭受损伤、dna复制和细胞分裂被阻止时被诱导的一种细胞自我保护措施;(5)reca:sos反应的诱导涉及40多个独立的sos基因,其中大部分编码参与dna保护、修复、复制、突变和代谢的蛋白质,reca蛋白是其中一种起重要作用的蛋白质之一。对dna的修复和维持基因组完整性起到了至关重要的作用。在大肠杆菌中,重组酶reca与dna断裂处的单链dna形成核蛋白细丝,并寻找同源的双链dna作为断裂修复的模板;(6)同源重组:含有同源序列的dna分子之间或分子之内的重新组合。
42.实施例1
43.peccas9-2.0质粒及其构建方法和应用
44.一、peccas9-2.0质粒
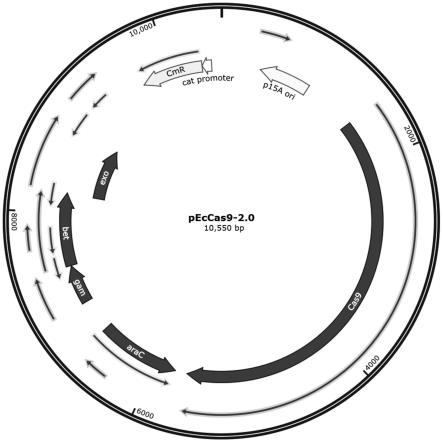
45.peccas9-2.0质粒的图谱结构如图1所示,图中黄色箭头为质粒的p15a复制子,cas9为表达cas9蛋白的基因,bet、gam和exonuclease为λ-red重组系统,能将cas9蛋白造成的基因组dna双链断裂通过同源重组的方式进行修复,该系统通过图谱中arac,即阿拉伯糖操纵子进行调控表达。cmr为氯霉素抗性基因,用于筛选质粒转化子。
46.具体的构建流程为:cas9基因及其启动子用引物p15a-cas9-up和p15a-cas9-dn扩增获得,所述p15a-cas9-up序列如seq id no.1所示,p15a-cas9-dn序列如seq id no.2所示。
47.seq id no.1:ttccaacagcttgaggcggaagctttaatgcgggtcctaacaccccttgtattactgt;
48.seq id no.2:cggtgatgccggccacgatgcgtccggcgtagaggatccactaaatcgatgcaggtggc。
49.然后将cas9基因及其启动子与线性化的p15a进行组装(组装方法参照南京诺维赞公司的clonexpress multis one step cloning kit试剂盒),获得质粒p15a-cas9,然后再在p15a-cas9的基础上叠加λ-red重组系统相关基因,获得peccas-2.0质粒。peccas-2.0质粒的配套grna质粒pecgrna来自于现有技术(li,q.,et al.,a modified pcas/ptargetf system for crispr-cas9-assisted genome editing in escherichia coli和jiang,yu,et al."multigene editing in the escherichia coli genome via the crispr-cas9 system.")。
50.实施例2
51.peccas9-2.0质粒在基因编辑中的应用
52.一、peccas9-2.0质粒能够作为基因编辑工具(利用peccas9-2.0质粒进行基因敲除)
53.以大肠杆菌mg1655为例,所述的基因敲除流程图如2所示,具体步骤如下:
54.1、查询序列信息,据此设计n20替换引物,同源引物及验证敲除引物;
55.2、根据现有记载构建靶向目标序列的pecgrna质粒并将构建产物用热激法转化到大肠杆菌dh5a中过夜培养;
56.3、抽提上一步得到的转化子的质粒,通过测序验证其n20序列部分是否与设计的相符,同时构建修复同源臂;
57.4、将peccas9-2.0质粒通过电转化的方式转化到大肠杆菌mg1655中过夜培养;
58.5、取上一步得到的转化子在液体培养基中过夜培养;
59.6、将pecgrna质粒和线性同源臂通过电转化的方式转化到含peccas9-2.0的大肠杆菌mg1655中过夜培养;
60.7、取上一步得到的转化子用验证引物通过菌落pcr验证敲除结果,验证弓|物设计在同源臂上下游约50bp处。
61.上述步骤中热激转化和电转化具体过程如下:
62.1、构建好的pecgrna质粒转化到大肠杆菌dh5α的热激转化方法:
63.(1)将感受态dh5α从-80℃冰箱中取出放到冰上解冻;
64.(2)在感受态细胞完全解冻后加入构建好的pecgrna质粒,吹打混匀后于冰上静置30min;
65.(3)将静置后的感受态细胞在42℃的温度下水浴60s,之后快速置于冰上冷却2min;
66.(4)加入900μl无抗生素的lb培养基,于37℃震荡复苏1h后在lb培养基平板上进行涂布,37℃过夜培养。
67.注意事项:
68.步骤(2)中,加入的构建产物的量不得超过感受态细胞总体积的十分之一。
69.步骤(3)中,冰上静置的过程中应避免震荡,否则会降低转化率。
70.本实验的pecgrna质粒含壮观霉素抗性基因,过夜培养时需要在培养基中加入终浓度100mg/l的壮观霉素进行筛选。
71.2、peccas-2.0质粒、pecgrna质粒及同源臂转化到大肠杆菌mg1655的电转化方法:
72.(1)将大肠杆菌mg1655菌株在lb培养基中在37℃,200rpm/min的条件下过夜培养;
73.(2)第二天按1%的接种量接种到50ml lb培养基中;
74.(3)当新接种的菌液生长到od
600
值为0.6~0.8时,将菌液冰浴20分钟充分冷却;
75.(4)将菌液置入50ml离心管中在4000rpm/min,4℃的条件下冷冻离心10分钟,将上清液倒出并将离心管迅速插回冰上;
76.(5)用冰预冷的无菌水或10%甘油溶液将沉菌在冰上吹打均匀,在4000rpm/min,4℃的条件下冷冻离心10分钟,将上清液倒出并将离心管迅速插回冰上;
77.(6)重复步骤(5)1-2次;
78.(7)根据转化样品数,按每份样品100μl的量向离心管中加入冰预冷的无菌去离子水,并与沉菌吹打混匀;
79.(8)按每份样品100μl的量将吹打好的菌液加入到插在冰上的1mm电转杯中;
80.(9)加入100ng需要转化的peccas-2.0质粒,混匀并确保菌液-质粒混合物处于电转杯内部的两个金属电极的中间;
81.(10)将装有菌液-质粒混合物的电转杯外侧金属电极仔细擦干后放入电转仪中在1.8kv、200ω、25μf条件下电击,电击后立刻加入900μl的无抗生素lb培养基并吹打均匀,之后将电转杯内的液体尽量吸入1.5ml离心管中;
82.(11)将装有电击后菌液的离心管37℃震荡复苏1h后在含氯霉素的lb培养基平板上进行涂布,37℃过夜培养;
83.(12)挑取转化子在含氯霉素的液体lb培养基中在37℃,200rpm/min的条件下过夜培养;
84.(13)第二天按1%的接种量接种到含有氯霉素的50ml lb培养基中并添加终浓度为10mm的l-阿拉伯糖;
85.(14)当新接种的菌液生长到od
600
值为0.6~0.8时,将菌液冰浴20分钟充分冷却;
86.(15)将菌液置入50ml离心管中在4000rpm/min,4℃的条件下冷冻离心10分钟,将上清液倒出并将离心管迅速插回冰上;
87.(16)用冰预冷的无菌dd水或10%甘油溶液将沉菌在冰上吹打均匀,在4000rpm/min,4℃的条件下冷冻离心10分钟,将上清液倒出并将离心管迅速插回冰上;
88.(17)重复步骤(16)1-2次;
89.(18)根据转化样品数,按每份样品100μl的量向离心管中加入冰预冷的无菌去离子水,并与沉菌吹打混匀;
90.(19)按每份样品100μl的量将吹打好的菌液加入到插在冰上的1mm电转杯中;
91.(20)加入100ng通过验证的pecgrna质粒和200-400ng同源臂,在总量不超过10μl的情况下混匀并确保菌液-质粒混合物处于电转杯内部的两个金属电极的中间;
92.(21)将装有菌液-质粒混合物的电转杯外侧金属电极仔细擦干后放入电转仪中在1.8kv、200ω、25μf条件下电击,电击后立刻加入900μl的无抗生素lb培养基并吹打均匀,之后将电转杯内的液体尽量吸入1.5ml离心管中;
93.(22)将装有电击后菌液的离心管37℃震荡复苏1h后在含氯霉素和壮观霉素的lb培养基平板上进行涂布,37℃过夜培养。
94.注意事项:
95.步骤(5)到步骤(9)及步骤(16)到步骤(20)的过程中尽量使沉菌或菌液保持冰浴状态。
96.电转时应避免在菌液-质粒混合物体系中残留有任何金属离子,否则可能会使电转失败。
97.培养时需要在培养基中的氯霉素和壮观霉素的终浓度分别为25mg/l和100mg/l。
98.二、实验结果
99.按照图2的流程获得peccas-2.0质粒在大肠杆菌中的逃逸率和编辑效率;
100.某位点的转化率计算方法为1微克dna经转化后得到的阳性克隆菌落数。
101.以control组为本次实验的空白对照,该组转化的pecgrna质粒不靶向任何位点,因此不会造成大肠杆菌基因组的断裂,也不会导致大肠杆菌死亡,仅用于计算当次实验的转化率;
102.计算peccas-2.0质粒在16个靶向大肠杆菌mg1655菌株染色体不同位置的pecgrna质粒的引导下,在不添加修复同源臂时所造成的逃逸率。
103.某位点的逃逸率计算方法为:逃逸率=靶向某位点的pecgrna质粒的转化率/本次实验做的空白对照pecgrna质粒的转化率。
104.利用转化子用验证引物通过菌落pcr验证敲除结果,五角星标记代表经过pcr验证,确定编辑成功的菌落,验证了peccas9-2.0质粒在大肠杆菌mg1655的cada和maeb基因中,在大肠杆菌bl21(de3)菌株的,cada、maeb、cynt基因中和大肠杆菌w菌株的cada、maeb基
因中的编辑率。具体结果如下:
105.1、peccas-2.0质粒在大肠杆菌中的逃逸率
106.如图3所示,peccas-2.0质粒在大肠杆菌中有较低的逃逸率,图3为peccas-2.0质粒在16个靶向大肠杆菌mg1655菌株染色体不同位置的pecgrna质粒的引导下,在不添加修复同源臂时所造成的逃逸率。
107.可以看出,在大多数基因位点上,本发明所使用的peccas9-2.0质粒在大多数位点都能够达到低于10-8
的逃逸率,符合预期目标。
108.2、peccas-2.0质粒在大肠杆菌中的编辑效率
109.peccas9-2.0质粒在大肠杆菌mg1655的cada和maeb基因中能够达到100%的编辑率(图4)。此外,peccas9-2.0质粒在大肠杆菌的其他菌株中也同样表现良好,在大肠杆菌bl21(de3)菌株的,cada、maeb、cynt基因(图5)以及大肠杆菌w菌株的cada、maeb基因中的编辑效率都达到了100%(图6)。
110.尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
111.显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1