一种恩扎卢胺中间体的制备方法与流程
1.本发明涉及一种恩扎卢胺中间体的制备方法,属于有机合成技术领域。
背景技术:
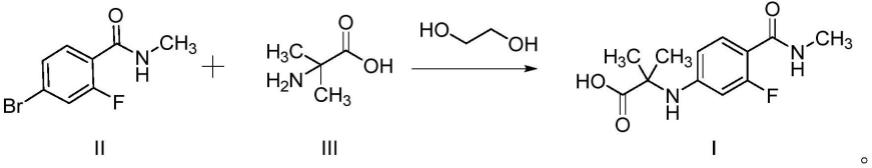
2.恩扎卢胺是用于治疗已扩散或复发的晚期去势前列腺癌的雄激素受体抑制剂药物。恩扎卢胺由安斯泰来和medivation共同开发,于2012年8月获得fda批准,商品名为xtandi。n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸是合成恩扎卢胺的关键中间体。
[0003]
专利w02011106570、cn103108549a公开了一种合成n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的方法:在2-乙酰基环己酮存在下,将化合物ii和化合物iii在dmf溶剂中进行偶联反应,得到n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸(即化合物i),其合成路线如下所示:
[0004][0005]
但是,该方法反应时间较长,在后处理中使用了化合物ii的16倍体积的水,极大的增加了生产中的废水量,并且析晶条件苛责,产物收率较低。
[0006]
专利cn105985258a报道了另外一种合成n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的方法:在1-(2,6,6-三甲基-1-环己烯基)丁烷-1,3-二酮存在下,将化合物ii和化合物iii在dmf溶剂中进行偶联反应,得到n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸,其合成路线如下所示:
[0007][0008]
虽然该方法将收率提高到了90%-92%,但是其同样采用dmf作为溶剂,存在以下问题:(1)生成的产物溶解在dmf中,后处理过程中需要再将dmf与产物分离,或萃取或蒸馏,操作繁琐;(2)由于碱不溶于dmf,因此该反应属于两相反应,为使反应完全则需要延长反应时间或提高反应温度,而这两种方式均会产生负面效果:a)如果延长反应时间,在碱性条件、100℃以上,dmf存在分解生成二甲胺杂质的可能,导致产品颜色较深,增加了化合物i及后续产品恩扎卢胺的杂质控制风险;b)如果提高反应温度,在较高温度下(110℃以上),化
合物i会发生如下降解,生成的降解杂质iv含有基因毒性警示结构,而且后续会参与反应,生成一系列的衍生杂质,难以清除,严重影响产品质量。
[0009][0010]
为了解决上述问题,亟需开发出一种高纯度、高收率、反应可控、后处理操作简便、三废少且绿色环保的方法来制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸。
技术实现要素:
[0011]
针对现有技术存在的问题,本发明提供了一种新的制备恩扎卢胺中间体—n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的方法。
[0012]
本发明具体提供了一种n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备方法,包括以下步骤:以化合物ii和化合物iii为原料,在乙二醇、卤化亚铜和无机碱的存在下,于非极性溶剂中反应,得到化合物i,即n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸;
[0013][0014]
进一步地,所述非极性溶剂为甲苯、乙苯、二甲苯中的一种或两种以上的混合物;
[0015]
和/或,所述的卤化亚铜为氯化亚铜、碘化亚铜、溴化亚铜中的一种或两种以上的混合物;
[0016]
和/或,所述无机碱为碳酸钾、碳酸钠、碳酸氢钠、碳酸铯中的一种或两种以上的混合物。
[0017]
进一步地,所述非极性溶剂为甲苯;
[0018]
和/或,所述的卤化亚铜为氯化亚铜;
[0019]
和/或,所述无机碱为碳酸钾。
[0020]
进一步地,所述化合物iii与化合物ii的摩尔比为(1.0~3.0):1;和/或,所述乙二醇与化合物ii的摩尔比为(0.05~1.0):1;
[0021]
和/或,所述非极性溶剂与化合物ii的体积质量比为3:1ml/g~15:1ml/g。
[0022]
进一步地,所述化合物iii与化合物ii的摩尔比为(1.5~1.8):1;
[0023]
和/或,所述乙二醇与化合物ii的摩尔比为(0.1~0.4):1;
[0024]
和/或,所述非极性溶剂与化合物ii的体积质量比为5:1ml/g~10:1ml/g。
[0025]
进一步地,所述无机碱与化合物ii的摩尔比为(2.0~5.0):1;
[0026]
和/或,所述卤化亚铜与化合物ii的摩尔比为(0.1~0.5):1。
[0027]
进一步地,所述无机碱与化合物ii的摩尔比为(2.5~3.0):1;
[0028]
和/或,所述卤化亚铜与化合物ii的摩尔比为(0.2~0.25):1。
[0029]
进一步地,所述反应的温度为80℃~130℃,反应的时间为3h~18h。
[0030]
进一步地,所述反应的温度为100℃~110℃,优选为105℃~110℃.
[0031]
进一步地,所述反应的时间为5h~15h,优选为5h~8h。
[0032]
进一步地,所述反应是在惰性气体的氛围下进行的。
[0033]
进一步地,所述惰性气体是氦气、氩气、氮气、氖气中的一种或两种以上的混合物。
[0034]
与现有技术相比,本发明制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的方法取得了以下有益效果:
[0035]
1.与公开号为cn103108549a(原研)的制备方法相比,本发明的方法缩短了反应时间,提高了转化率,降低了反应溶剂体积和后处理中水的体积,收率明显提高。
[0036]
2.与公开号为cn105985258a的制备方法相比,本发明的方法缩短了反应时间,无过滤无机盐、蒸馏dmf等步骤,无打浆操作,大大简化了后处理操作,降低了操作成本,产品的性状明显改善,纯度和收率也有提高。
[0037]
3.本发明的制备方法中,乙二醇既是配体又是相转移催化剂。在反应中,乙二醇的加入大大提高了催化剂的活性,同时,乙二醇具有相转移催化剂的特性,使催化剂与底物更有效地发生分子碰撞,从而使反应更容易进行,提高了反应速率。本发明的制备方法中,采用的反应溶剂为非极性溶剂,生成的产物可用少量的水萃取,简化了操作并大大降低了废水量,实现了三废小、绿色环保的目标。
[0038]
本发明的制备方法将多功能性的配体和非极性溶剂结合,减少了反应时间,提高了反应转化率,降低了溶剂用量,简化了处理过程,实现了高收率、高纯度、杂质少、反应可控的目标,具有成本低、三废小、绿色环保的优势,应用前景广阔。
[0039]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
附图说明
[0040]
图1为实施例1中合成得到的n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的1h-nmr图谱。
[0041]
图2为实施例1中合成得到的n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的ms图谱。
具体实施方式
[0042]
如无特别说明,本发明具体实施方式中所用试剂和原料均可通过购买市售产品获得。
[0043]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0044]
实施例1:n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备
[0045][0046]
在3l三口瓶中加入化合物ii(200.00g,0.86mol),化合物iii(133.32g,1.29mol),碳酸钾(297.80g,2.15mol),乙二醇(21.40g,0.34mol),甲苯1l,置换氮气三次,再加入氯化亚铜(17.07g,0.17mol),再次置换氮气三次,升温至105℃,5h后tlc监测,化合物ii完全消失,停止反应,冷却至室温。
[0047]
向反应瓶中加水1l,搅拌使全溶,分相得水相,再加入二氯甲烷400ml萃取,除去反应液中有机杂质。水相在搅拌下,缓慢倾到3mol/l盐酸溶液,调节反应液体系ph至3.0~3.5,然后冷却至10℃以下析晶1h。过滤,55℃下鼓风干燥12h,得到白色固体213.2g,收率97.3%,hplc纯度99.7%。
[0048]
对本实施例得到的产品进行1h-nmr和ms分析,如图1、2所示。
[0049]1h-nmr(dmso-d6,400mhz):δ1.44(s,6h),δ2.71~2.73(d,3h),δ6.13~6.17(dd,1h),δ6.32~6.35(dd,1h),δ7.42~7.47(t,1h),δ7.64~7.66(t,1h);ms:253.3(m-h
+
)。
[0050]
实施例2:n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备
[0051]
在5l三口瓶中加入化合物ii(200.00g,0.86mol),化合物iii(133.32g,1.29mol),碳酸钾(297.80g,2.15mol),乙二醇(21.40g,0.34mol),甲苯2l,置换氮气三次,再加入氯化亚铜(17.07g,0.17mol),再次置换氮气三次,升温至105℃,5h后tlc监测,化合物ⅱ完全消失,停止反应,冷却至室温。
[0052]
向反应瓶中加水2l,搅拌使全溶,分相得水相,再加入二氯甲烷800ml萃取,除去反应液中有机杂质。水相在搅拌下,缓慢倾到3mol/l盐酸溶液,调节反应液体系ph至3.0~3.5,然后冷却至10℃以下析晶1h。过滤,55℃下鼓风干燥12h,得到白色固体212.8g,收率97.1%,hplc纯度99.8%。
[0053]
实施例3:n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备
[0054]
在3l三口瓶中加入化合物ii(200.00g,0.86mol),化合物iii(133.32g,1.29mol),碳酸钾(297.80g,2.15mol),乙二醇(5.35g,0.09mol),甲苯1l,置换氮气三次,再加入氯化亚铜(17.07g,0.17mol),再次置换氮气三次,升温至105℃,5h后tlc监测,化合物ⅱ未完全消失,继续反应,3h后再次tlc监测,化合物ⅱ完全消失,冷却至室温。
[0055]
向反应瓶中加水1l,搅拌使全溶,分相得水相,再加入二氯甲烷400ml萃取,除去反应液中有机杂质。水相在搅拌下,缓慢倾到3mol/l盐酸溶液,调节反应液体系ph至3.0~3.5,然后冷却至10℃以下析晶1h。过滤,55℃下鼓风干燥12h,得到白色固体204.2g,收率93.2%,hplc纯度99.6%。
[0056]
实施例4:n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备
[0057]
在3l三口瓶中加入化合物ii(200.00g,0.86mol),化合物iii(133.32g,1.29mol),碳酸钾(297.80g,2.15mol),乙二醇(21.40g,0.34mol),甲苯1l,置换氮气三次,再加入氯化亚铜(17.07g,0.17mol),再次置换氮气三次,升温至105℃,15h后tlc监测,化合物ⅱ完全消失,停止反应。
[0058]
向反应瓶中加水1l,搅拌使全溶,分相得水相,再加入二氯甲烷400ml萃取,除去反应液中有机杂质。水相在搅拌下,缓慢倾到3mol/l盐酸溶液,调节反应液体系ph至3.0~3.5,然后冷却至10℃以下析晶1h。过滤,55℃下鼓风干燥12h,得到白色固体212.5g,收率97.0%,hplc纯度99.5%。
[0059]
表1.实施例1~4和对比例1的反应条件、产物纯度及收率
[0060][0061][0062]
比较表1中实施例1和实施例2的数据可以看出,增大反应溶剂量,对于产物收率和纯度基本无影响,说明产物易于用水萃取,在溶剂中无残留。
[0063]
比较表1中实施例1和对比例1的数据可以看出,不使用乙二醇时,原料(化合物ii)不能反应完全,收率大幅降低,纯度低;比较表1中实施例3和对比例1的数据可以看出,使用0.1当量的乙二醇,则反应时间大幅缩短,原料反应完全,收率大幅提高,纯度高,说明乙二醇能够促进反应转化,提高反应效率。
[0064]
比较表1中实施例1和实施例4的数据可以看出,延长反应时间,产物纯度及性状均正常,克服了现有技术中以dmf为溶剂时,在延长反应时产物降解成杂质的问题。说明使用甲苯做溶剂、乙二醇做配体能降低杂质风险。
[0065]
以下为制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的对比例。
[0066]
对比例1:n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸的制备在3l三口瓶中加入化合物ii(200.00g,0.86mol),化合物iii(133.32g,1.29mol),碳酸钾(297.80g,2.15mol),甲苯1l,置换氮气三次,再加入氯化亚铜(17.07g,0.17mol),再次置换氮气三次,升温至105℃,15h后tlc监测,化合物ⅱ未完全消失,冷却至室温。
[0067]
向反应瓶中加水1l,搅拌使全溶,分相得水相,再加入二氯甲烷400ml萃取,除去反应液中有机杂质。水相在搅拌下,缓慢倾到3mol/l盐酸溶液,调节反应液体系ph至3.0~3.5,然后冷却至10℃以下析晶1h。过滤,55℃下鼓风干燥12h,得到白色固体84.6g,收率38.6%,hplc纯度85.8%。
[0068]
对比例2:公开号为cn103108549a(原研)的制备方法
[0069]
参照公开号为cn103108549a的中国专利记载的方法,制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸,具体操作如下:往烧瓶中加入溴苯甲酰胺(化合物ii)(10g,43.1mmol)、氨基丙丁酸(化合物iii)(6.7g,64.6mmol,1.5当量)、k2co3(15g,2.5当量)、dmf(60ml,6体积)和水(1.8ml),并将反应浆液加热至30℃。向反应浆液中加入2-乙酰基环己酮(1.14ml,8.1mmol,0.2当量),然后在氮气下105℃搅拌14小时。hplc分析显示96.6%转变为目标产物。随后将反应混合物冷却至室温(rt)并用水(120ml)和ipac(60ml)进行萃取。下层水层采用ipac(60ml)再次萃取后用180ml1m柠檬酸酸化至ph为4.0。产物在室温下开始结晶并将该批进一步冷却至5-7℃、过滤、水洗(40ml)并在真空下50℃干燥12小时。反应收获8.0g黄褐色固体状的产物(73.0%收率),hplc纯度为99.4%。
[0070]
对比例3:公开号为cn105985258a的制备方法
[0071]
参照公开号为cn105985258a的中国专利申请记载的方法,制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸,具体操作如下:在3l的三颈瓶中加入n-甲基-4-溴-2-氟-苯甲酰胺(化合物ii)(200g,0.86mol),2-氨基异丁酸(化合物iii)(133g,1.29mol),氯化亚铜(16.8g,0.17mol),化合物a(1-(2,6,6-三甲基-1-环己烯基)丁烷-1,3-二酮)(20.8g,0.1mol)以及碳酸钾固体(303.6g,2.2mo1),然后加入1l的dmf和20ml的蒸馏水,置换氮气三次。开启加热升温到115℃,tlc检测,10个小时后,原料完全消失。停止反应。冷却到室温。
[0072]
过滤,除去固体不溶物,滤渣再用少许dmf洗涤,合并滤液,减压蒸干,加入400ml水使体系完全溶解,用400ml醋酸异丙酯萃取2次,除去残液的有机杂质。水相在搅拌下,向其中滴加饱和柠檬酸溶液,调节反应体系的ph=4-5,然后冷却到10℃以下,有大量浅绿色固体析出。过滤,滤饼用800ml水打浆30分钟,过滤;滤饼再用200ml的醋酸异丙酯打浆1小时,过滤,60度真空干燥至恒重,得到黄褐色固体199.4g,收率91%,hplc纯度99.2%。
[0073]
比较实施例1与对比例2可以看出,采用本发明的方法制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸,反应时间短,转化率高,降低了反应溶剂体积和后处理中水的体积,收率明显提高。
[0074]
比较实施例1与对比例3可以看出,采用本发明的方法制备n-[3-氟-4-[(甲基氨基)羰基]苯基]-2-甲基丙氨酸,反应时间短,无过滤无机盐和蒸馏dmf等步骤,无打浆操作,大大简化了后处理操作,降低了操作成本,产品的性状明显改善,纯度和收率也有提高。
[0075]
综上,本发明提供了一种恩扎卢胺中间体的制备方法。本发明的制备方法将多功能性的配体和非极性溶剂结合,减少了反应时间,提高了反应转化率,降低了溶剂用量,简化了处理过程,实现了高收率、高纯度、杂质少、反应可控的目标,具有成本低、三废小、绿色环保的优势,应用前景广阔。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1