一种3-乙酰基-2-氯吡啶的合成方法与流程
羟基吡啶的酯分子量较大,该路线的原子经济性不高,而且合成酯的原料很贵,生产成本高,不适合放大生产。
9.2010年,英国爱丁堡大学的mcnab hamish和gaywood alexander p.报道(organic and biomolecular chemistry, 2010, 8, 5166-5173),2-氯烟腈和4.27个当量的甲基氯化镁反应,后处理后得到3-乙酰基-2-氯吡啶,收率65%。2-氯烟腈成本较高,且需要用到过量的格式试剂,不适合放大生产。
10.除了可以与镁试剂和锂试剂反应外,3-乙酰基-2-氯吡啶还可由烟酰氯与四甲基锡反应合成。1997年,美国syntex inc公司在专利us5688795中报道,在双(苯腈)二氯化钯(ii)催化下2-氯烟酰氯与四甲基锡反应,经硅胶柱层析纯化得到3-乙酰基-2-氯吡啶,收率54%。收率不高,原料四甲基锡不易得、成本高,而且还需要用到昂贵的金属钯催化剂,也不适合工业化生产。
11.除了如上所述通过与有机金属试剂反应合成3-乙酰基-2-氯吡啶外,还可以通过烟酰氯与丙二酸二乙酯酰化、脱羧合成3-乙酰基-2-氯吡啶。2007年,memory pharmaceuticals公司在美国专利us20070078147中报道,2-氯烟酸和草酰氯反应生成2-氯烟酰氯,收率98%;在无水氯化镁催化下2-氯烟酰氯和丙二酸二乙酯反应生成2-(2-氯烟酰基)丙二酸二乙酯,收率85%;在湿的dmso中2-(2-氯烟酰基)丙二酸二乙酯在130℃脱羧、经硅胶柱层析纯化得产品,收率52%。三步反应总收率43.3%,总收率不高。
12.路线二、以3-乙酰基吡啶为原料。
13.2010年,英国爱丁堡大学的mcnab hamish和gaywood alexander p.报道(organic and biomolecular chemistry, 2010, 8, 5166
ꢀ‑ꢀ
5173),3-乙酰基吡啶在30%双氧水氧化下生成3-乙酰基吡啶n-氧化物,然后与三氯氧磷在100℃反应1小时,后处理得到3-乙酰基-2-氯吡啶,两步反应总收率33%。总收率不高,且原料3-乙酰基吡啶价格较贵,生产成本高,不合放大生产。
14.路线三、以2-氯吡啶为原料。
15.2011年,美国堪萨斯大学在专利wo2011005759中报道,以新制备的lda作碱,2-氯吡啶和乙醛进行加成反应,经后处理、硅胶柱层析纯化后得到1-(2-氯吡啶-3-基)乙醇,然后用三氧化铬氧化得到乙酰基-2-氯吡啶粗品,硅胶柱层析纯化后得到纯品。两步反应总收率51.8%。总收率不高,需要零下78℃的低温,还需要硅胶柱层析纯化,不适合工业化生产。
16.总之,现有的工艺存在着或多或少的缺点:或者原材料成本高,或者总收率低,或者反应生产周期长,或者需要柱层析纯化等不适合放大的操作步骤等,都不适合工业化生产。为了提供大量的、价廉的3-乙酰基-2-氯吡啶产品,为后续药物开发提供保障,非常有必要开发一条新的合成该化合物的方法。
技术实现要素:
17.针对现有工艺存在着或者成本高、或者收率低、或者反应生产周期长、或者需要柱层析纯化,都不适合工业化生产缺点。本发明开发出了一条原料价廉易得、反应安全易操作、反应条件温和、且反应后处理简单、收率较高,适合放大生产,具有显著工业价值的合成方法。
18.为了实现上述目的,本发明提供一种3-乙酰基-2-氯吡啶的合成方法,所述方法包括:以2-氯烟酸为原料,与含锂化合物反应生成2-氯烟酸锂盐,2-氯烟酸锂盐经干燥后与甲基溴化镁进行加成反应,生成3-乙酰基-2-氯吡啶;所述含锂化合物为氢氧化锂一水合物和/或锂盐。
19.根据本发明一种优选实施方式,生成2-氯烟酸锂盐的步骤包括:以水为溶剂,使2-氯烟酸和含锂化合物接触进行反应,得到2-氯烟酸锂盐。
20.进一步地,所述2-氯烟酸与含锂化合物的摩尔比为0.9~1.0:1。
21.进一步地,生成2-氯烟酸锂盐的反应温度为40~50℃。
22.根据本发明,所述锂盐优选为碳酸锂。
23.根据本发明一种优选实施方式,2-氯烟酸锂盐与甲基溴化镁进行加成反应的步骤包括:(1)在惰性气体保护下,将第一有机溶剂、2-氯烟酸锂盐搅拌混合,然后滴加甲基溴化镁,滴毕自然升温至加成反应温度进行反应,得到反应液;(2)在惰性气体保护下,将反应液滴加至低温水中,搅拌第一时间后,滴加盐酸,滴毕升温至20~25℃,搅拌第二时间,静置分液,用第二有机溶剂萃取水相后,合并有机相,浓
缩得到目标产物。
24.进一步地,步骤(1)中,所述第一有机溶剂选自四氢呋喃和2-甲基四氢呋喃中的至少一种。
25.进一步地,步骤(1)中,所述2-氯烟酸锂盐与甲基溴化镁的摩尔比为1:1~1.5,优选为1:1.1~1.3。
26.进一步地,步骤(1)中,相对每1000kg反应体系,所述甲基溴化镁的滴加速度为0.9~1.8kg/min。
27.进一步地,步骤(1)中,滴加甲基溴化镁前控制体系温度至-4℃以下,滴加过程中控制体系温度不超过0℃。
28.进一步地,步骤(1)中,所述加成反应温度为12~18℃,反应时间为0.5-1.5h。
29.进一步地,步骤(2)中,所述低温水的温度为4℃以下。
30.进一步地,步骤(2)中,相对于1kg反应液,所述低温水的质量为0.3~0.5kg。
31.进一步地,步骤(2)中,滴加过程中控制体系温度不超过15℃。
32.进一步地,步骤(2)中,所述第一时间为0.5~1.5h。
33.进一步地,步骤(2)中,所述盐酸的摩尔加入量为2-氯烟酸锂盐摩尔投料量的1.8~2.0倍。
34.进一步地,步骤(2)中,所述第二时间为1.5~2.5h。
35.进一步地,步骤(2)中,所述第二有机溶剂选自乙酸乙酯、二氯甲烷、甲基叔丁基醚中的至少一种。
36.本发明各步骤中所述惰性气体可以为氮气。
37.根据本发明,需控制进行步骤(2)的时间点,具体地,判断进行步骤(2)的方法和标准为:通过hplc检测反应结果,体系中2-氯烟酸小于21%,进行步骤(2)。
38.本发明的方法中,需对反应生成2-氯烟酸锂盐进行干燥再进行后续的加成反应,具体地,需将2-氯烟酸锂盐干燥至含水量小于1%。
39.本发明的合成方法还包括精馏步骤:精馏塔中加入3-乙酰基-2-氯吡啶粗品,开加热,开油泵拉干低沸物,切换至罗茨泵,气相温度逐渐升高至有前馏份蒸出,定期取样检测,当最大杂质小于0.5%时,切换至主馏份收集罐,定期取样检测,当主峰后杂质大于0.5%时停止精馏,得到3-乙酰基-2-氯吡啶产品。
40.本发明的合成方法具有以下优势:(1)反应收率高,产品纯度大于99%,单杂小于0.5%。
41.(2)通过加入价廉的含锂化合物如氢氧化锂一水合物形成锂盐,可节省1当量甲基溴化镁,反应经济性好。
42.(3)反应条件温和、反应安全易操作、且反应后处理简单(无需柱层析)、适合放大生产,具有显著工业价值。
43.本发明的其它特征和优点将在随后具体实施方式部分予以详细说明。
附图说明
44.通过结合附图对本发明示例性实施方式进行更详细的描述,本发明的上述以及其它目的、特征和优势将变得更加明显。
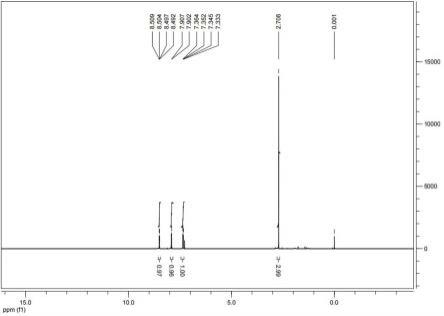
45.图1为本发明实施例1合成的3-乙酰基-2-氯吡啶的核磁图谱。
46.图2为本发明实施例1合成的3-乙酰基-2-氯吡啶的gc图。
47.图3为本发明实施例2合成的3-乙酰基-2-氯吡啶的gc图。
具体实施方式
48.下面将更详细地描述本发明的优选实施方式。虽然以下描述了本发明的优选实施方式,然而应该理解,可以以各种形式实现本发明而不应被这里阐述的实施方式所限制。
49.实施例12-氯烟酸锂盐的合成:向1000l反应釜中加入自来水325kg,开动搅拌,加入氢氧化锂一水合物171.8kg(1eq.)。打开冷盐水,向反应釜中快速加入2-氯烟酸600kg,控温40~50℃反应2h,控制40℃放料,降温至20℃以下,抽滤,烘箱烘干至含水量小于1%,得产品2-氯烟酸锂盐660kg,收率98%。
50.乙酰基-2-氯吡啶的合成:向2000l反应釜中加入四氢呋喃200kg,开动搅拌,加入2-氯烟酸锂盐100kg,然后加入四氢呋喃250kg,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁300kg(1.2eq.),控制滴加速度(前20% 0.6kg/min,后80% 0.8kg/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余19.9%,双甲基杂质含量1.3%,产品含量78.5%。向另一3000l反应釜中加入水350kg,氮气保护下降温至0℃左右,滴加2000l反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸182l,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯200l萃取一次,合并有机相,80℃以下浓缩至不出馏分。再减压精馏得产品73.5kg,收率:73.8%,gc纯度:99.5%。图1为实施例1合成的3-乙酰基-2-氯吡啶的核磁图谱。图2为实施例1合成的3-乙酰基-2-氯吡啶的gc图。
51.精馏方法:精馏塔中加入3-乙酰基-2-氯吡啶粗品,开加热,开油泵拉干低沸物,液相温度升至80℃左右有馏份蒸出,当液相温度升至120℃,气相温度升至80℃无明显馏份蒸出。切换至罗茨泵,气相温度逐渐升高至130℃左右,液相温度140℃左右,有前馏份蒸出,每20分钟取样检测一次,最大杂质小于0.5%,切换至主馏份收集罐(气相温度逐渐降至116℃,液相温度降至124℃),前馏份单独放置。每隔1h gc监控一次,主峰后杂质逐渐增大,当主峰后杂质大于0.5%时停止精馏。
52.实施例2
2-氯烟酸锂盐的合成:同实施例1。
53.乙酰基-2-氯吡啶的合成:向200l反应釜中加入2-甲基四氢呋喃20kg,开动搅拌,加入2-氯烟酸锂盐10kg,然后加入2-甲基四氢呋喃25kg,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30kg(1.2eq.),控制滴加速度(0.5kg/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余19.8%,双甲基杂质含量1.5%,产品含量78.3%。向另一300l反应釜中加入水35kg,氮气保护下降温至0℃左右,滴加200l反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18l,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20l萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品6.7kg,收率:69.3%,gc纯度:99.2%。图3为实施例2合成的3-乙酰基-2-氯吡啶的gc图。
54.实施例32-氯烟酸锂盐的合成:向300ml反应釜中加入自来水200g,向反应釜中快速加入2-氯烟酸60g,开动搅拌,控温40~50℃,加入氢氧化锂一水合物17.2g(1eq.)的水溶液50g,反应2h,打开冷盐水,降温至20℃以下,抽滤,烘箱烘干至含水量小于1%,得产品2-氯烟酸锂盐61g,收率98%。
55.乙酰基-2-氯吡啶的合成:向200ml反应瓶中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸锂盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液25g(1.0eq.),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余28.6%,双甲基杂质含量1.5%,产品含量69.6%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸15ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品6.1g,收率:63.1%,gc纯度:99.4%。
56.实施例42-氯烟酸锂盐的合成: 同实施例3。
57.乙酰基-2-氯吡啶的合成:向200ml反应瓶中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸锂盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮
气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30g(1.2eq.),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余18.3%,双甲基杂质含量1.6%,产品含量79.8%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品7.2g,收率:74.5%,gc纯度:99.5%。
58.实施例52-氯烟酸锂盐的合成:同实施例3。
59.乙酰基-2-氯吡啶的合成:向200ml反应瓶中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸锂盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液37.5g(1.5eq),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余49.3%,双甲基杂质含量3.5%,产品含量46.7%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸24ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品7.0g,收率:72.4%,gc纯度:99.1%。
60.实施例62-氯烟酸锂盐的合成:同实施例3。
61.乙酰基-2-氯吡啶的合成:向200ml反应瓶中加入2-甲基四氢呋喃20g,开动搅拌,加入2-氯烟酸锂盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30g(1.2eq),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余20.1%,双甲基杂质含量1.6%,产品含量77.9%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品7.1g,收率:73.5%,gc纯度:
99.5%。
62.实施例72-氯烟酸锂盐的合成:向300ml反应釜中加入自来水100g,碳酸锂14g(0.5eq.),向反应釜中快速加入2-氯烟酸60g,开动搅拌,控温80℃,反应2h,打开冷盐水,降温至20℃以下,抽滤,烘箱烘干得产品2-氯烟酸锂盐55.8g,收率89.8%。
63.乙酰基-2-氯吡啶的合成:向200ml反应釜中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸锂盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30g(1.2eq.),控制滴加速度(0.5g/min),滴加温度10~15℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余19.2%,双甲基杂质含量1.7%,产品含量78.8%,停止反应。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品7.1g,收率:73.7%,gc纯度:99.1%。
64.对比例12-氯烟酸钠盐的合成:向300ml反应釜中加入自来水100g,氢氧化钠15.2g(1eq.),向反应釜中加入2-氯烟酸60g,开动搅拌,控温40~50℃,反应2h,减压蒸馏干,然后加入150g的甲醇,降温至20℃以下,抽滤,烘箱烘干得产品2-氯烟酸钠盐60g,收率87.7%。
65.以2-氯烟酸钠盐合成3-乙酰基-2-氯吡啶:向200ml反应釜中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸钠盐11g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30g(1.2eq),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余38.8%,双甲基杂质含量3.2%,产品含量56.4%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品4.9g,收率:50.8%,gc纯度:98.1%。
66.对比例22-氯烟酸钠盐的合成:向300ml反应釜中加入自来水100g,碳酸钠20g(0.5eq.),向反应釜中快速加入2-氯烟酸60g,开动搅拌,控温40~50℃,反应2h,打开冷盐水,降温至20℃以下,抽滤,烘箱烘干得产品2-氯烟酸钠盐61.5g,收率90%。
67.乙酰基-2-氯吡啶的合成:向200ml反应釜中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸钠盐10g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液37.5g(1.5eq.),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余32.1%,双甲基杂质含量4.1%,产品含量63.2%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸24ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品4.8g,收率:55%,gc纯度:98.5%。
68.对比例32-氯烟酸钾盐的合成:向300ml反应釜中加入自来水100g,氢氧化钾21.3g(1eq.),向反应釜中加入2-氯烟酸60g,开动搅拌,控温40~50℃,反应2h,减压蒸馏干,然后加入150g的甲醇,降温至20℃以下,抽滤,烘箱烘干得产品2-氯烟酸钾盐66g,收率88.6%。
69.以2-氯烟酸钾盐合成3-乙酰基-2-氯吡啶:向200ml反应瓶中加入四氢呋喃20g,开动搅拌,加入2-氯烟酸钾盐12g,然后加入四氢呋喃25g,氮气置换空气一次(釜内压力小于-0.08mpa方可置换氮气),打开放空阀,氮气保护下降温至-4℃以下。使用蠕动泵滴加甲基溴化镁的四氢呋喃溶液30g(1.2eq),控制滴加速度(0.5g/min),滴加温度不得超过0℃。滴毕自然升温至15℃,保温反应1h,得到反应液。hplc检测反应结果,2-氯烟酸剩余49.3%,双甲基杂质含量3.5%,产品含量46.7%。向另一300ml反应釜中加入水35g,氮气保护下降温至0℃左右,滴加200ml反应釜中反应液,滴加温度不得超过10℃。滴毕,搅拌1h。然后滴加6mol/l盐酸18ml,滴加温度不得超过10℃。滴毕,升温至20-25℃,搅拌2h,静置分液。水相用乙酸乙酯20ml萃取一次,合并有机相,80℃以下浓缩至不出馏分。再采用实施例1的方法减压精馏得产品3.8g,收率:39.3%,gc纯度:98.2%。
70.以上已经描述了本发明的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1