一种雷替曲塞的关键杂质及其制备方法与流程
一种雷替曲塞的关键杂质及其制备方法
1、技术领域
1.本发明属于药物合成技术领域,具体涉及一种雷替曲塞的关键杂质及其制备方法。
2、
背景技术:
2.雷替曲塞首先于1995年在英国批准,1996年3月率先在英国上市,用于晚期结直肠癌 (mcrc),商品名为剂型为冻干粉(2毫克)。随后,在法国、爱尔兰、比利时、西班牙、意大利、奥地利等14个欧盟国家获得批准。在欧盟以外国家,雷替曲塞于1996年 7月15日在澳大利亚批准,同年11月在加拿大批准;2000年在新加坡批准,2002年土耳其获得批准。该药物用于治疗结肠直肠癌的适应症已在20多个国家批准。在部分国家,如捷克共和国、匈牙利、奥地利、比利时、意大利、爱尔兰、土耳其和葡萄牙,雷替曲塞还被批准用于治疗恶性胸膜间皮瘤(与顺铂或奥沙利铂联合)。
[0003][0004]
杂质是影响药品质量的一个重要因素,药品中杂质控制对药品的安全有着重要意义, ich(人用药品注册技术要求国际协调会)对不同种类的杂质限量进行了详细的规定。在药品质量研究过程中,对于原料制备工艺中实际存在的杂质必须确认杂质的结构、确定其毒性并控制杂质的含量。在药品申报注册中,根据fda及cfda的要求必须对产品中的杂质进行严格的控制,对出现在最终产品的杂质必须进行结构分析及质量控制,其来源和去除需要在注册文件中进行描述。
[0005]
api的杂质主要来自api本身的降解和制造过程。工艺杂质包括未反应的原材料,中间体,反应副产物,原材料或中间体中所含的杂质的衍生物,反应过程使用的溶剂、试剂、催化剂残留或其参与反应的杂质,api降解产物等。
[0006]
在发明人的系列研究中发现,雷替曲塞中有一个关键未知杂质,在原料和市售制剂中均有检出,但现有文献中均未见报道。因此,进一步识别、研究和化学表征雷替曲塞的杂质对进一步提高雷替曲塞质量控制的水平和安全性有重要意义。
[0007]
针对现有技术的不足,本发明人对雷替曲塞该未知杂质进行了系统研究,经分离制备明确了该关键杂质结构,进而用于雷替曲塞的纯度、杂质和定量分析,以控制产品质量。
3、
技术实现要素:
[0008]
本发明的目的是提供一种雷替曲塞的关键杂质的结构,并提供一种简洁有效的制备方法,以满足药物质量控制的需要。
[0009]
本发明提供了一种雷替曲塞关键杂质化合物a,结构如下式所示。
[0010][0011]
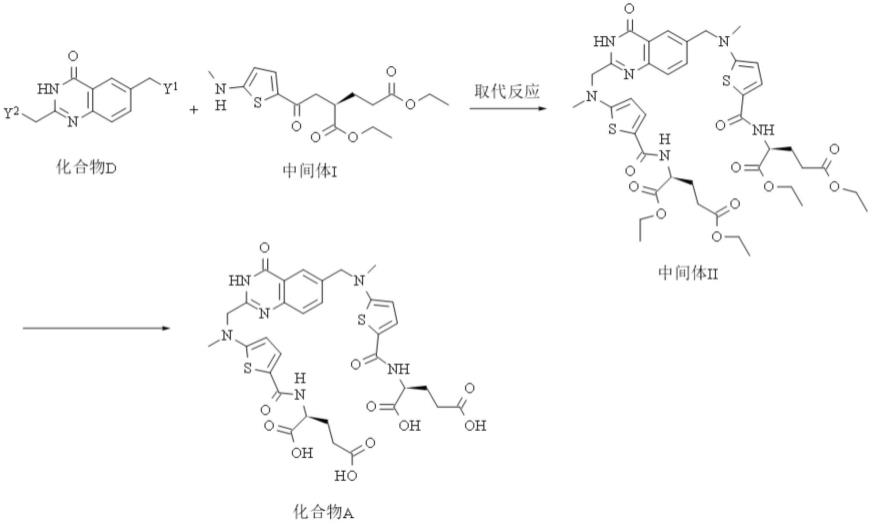
本发明还明确了关键杂质化合物a的产生机制,如下所示。本发明还制定了工艺控制策略,可以将化合物a有效的控制在合理的限度以下。已知的制备雷替曲塞的方法采用化合物c引入甲基喹唑啉酮片段,而化合物c则由化合物b经过卤代反应制备得到。研究中发现卤化过程不可避免会产生化合物d杂质,化合物d的性质与化合物c十分接近,一般生产工艺的纯化步骤无法将其完全清除。经过后续反应化合物d传递成为化合物a。这一杂质产生和传递过程已通过对不同路线制备得到的雷替曲塞,及其相关中间体的检测得到证实。
[0012][0013]
其中,所述的x1和x2分别选自卤素原子,所述的卤素原子为氟、氯、溴或碘。
[0014]
通过浓缩正常生产工艺的母液分离纯化可以得到化合物a,但由于其性质与主成分接近,含量偏低(<1%),获得满足对照品要求的足量化合物a,在有适合仪器设备支持条件下,还需要耗费较长时间和大量溶剂。
[0015]
本发明进一步提供了一种化合物a的制备方法,反应步骤为:
[0016]
(1)化合物d和中间体ⅰ在溶剂中进行反应,制备得到中间体ii;
[0017]
(2)中间体ⅱ溶解在溶剂中,加入碱进行水解反应,纯化后得到化合物a;
[0018][0019]
其中,所述的y1和y2相同或不同,分别选自卤素原子,所述的卤素原子为氟、氯、溴或碘,并且y1为溴时,y2不为氟或氯。优选的,y1和y2相同或不同,分别溴或碘。
[0020]
所述的步骤(1)中的溶剂包括但不限于n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、四氢呋喃、2-甲基四氢呋喃、丁酮、丙酮、n-甲基吡咯烷酮、环丁砜、三氯甲烷中的一种或几种,优选为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜等高极性溶剂的一种或几种。所述的反应温度为20-100℃,优选40-60℃。
[0021]
所述的步骤(2)中的溶剂包括但不限于水、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、四氢呋喃、2-甲基四氢呋喃、丁酮、丙酮、n-甲基吡咯烷酮、环丁砜、二氯甲烷、三氯甲烷、甲醇、乙醇、异丙醇中的一种或几种。所述的反应温度为10-90℃,优选20-50℃。
[0022]
所述的步骤(2)中的碱为水溶性金属氢氧化物、碳酸盐、磷酸盐、碱金属氧化物、碱土金属氧化物等,包括但不限于氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、氢氧化锂、碳酸锂、氢氧化镁、氢氧化钙、磷酸钠、磷酸钾、磷酸氢钾、磷酸氢镁等;优选氢氧化钠、氢氧化钾、氢氧化锂、碳酸钾、碳酸钠等常见无机碱或碱性无机盐。
[0023]
所述的步骤(2)中的纯化包括但不限于ph调节纯化、萃取洗涤纯化、硅胶色谱柱纯化及制备液相等仪器纯化的一种或多种合用。优选ph调节纯化、萃取洗涤纯化和硅胶色谱柱纯化。
[0024]
本发明还提供了一种化合物d的制备方法,反应步骤为:
[0025]
(1)氯乙腈和2-氨基-5-甲基苯甲酸经缩合反应制得化合物d-1;
[0026]
(2)化合物d-1与卤化试剂反应制得化合物d-2;
[0027]
(3)化合物d-2与卤化试剂反应制得化合物d;
[0028][0029]
其中,所述的y1和y2相同或不同,分别选自卤素原子,所述的卤素原子为氟、氯、溴或碘,并且y1为溴时,y2不为氟或氯。优选的,y1和y2相同或不同,分别溴或碘。
[0030]
所述的卤化试剂选自溴化钠、溴化钾、溴化锂、溴化镁、溴化铁、溴化亚铜、溴化镍、溴化铝、溴化锌、三溴化磷、五溴化磷、氢溴酸、碘化钾、碘化钠、碘化锂、碘化亚铜、溴素、溴水、n-溴代琥珀酰亚胺、二溴海因、n-溴代乙酰胺、碘、氯化碘、n-碘代琥珀酰亚胺、 n-碘代乙酰胺。所述的步骤(2)中卤化试剂优选为溴化钠、溴化钾、溴化锂、碘化钾或碘化亚铜。所述的步骤(3)中卤化试剂优选为溴素、溴水、n-溴代琥珀酰亚胺、二溴海因、n
‑ꢀ
溴代乙酰胺、碘、氯化碘、n-碘代琥珀酰亚胺或-碘代乙酰胺。
[0031]
本发明还提供了一种化合物d,结构式如下所示:
[0032][0033]
所述的y1和y2相同或不同,分别选自卤素原子,所述的卤素原子为氟、氯、溴或碘,并且y1为溴时,y2不为氟或氯。
[0034]
本发明的有益技术效果为:
[0035]
①
本发明提供了一种雷替曲塞的关键有关物质化合物a,确证了其结构,并提供了该关键杂质的简单、高效的制备方法,制备所得杂质纯度高;
[0036]
②
原料药生产厂商可以根据其结构明确其产生机制及产生的影响因素,并在生产工艺中制定合理的控制策略,优化检测方法,确保产品质量及安全有效;
[0037]
③
可以较好的满足雷替曲塞质量研究与控制对关键杂质对照品化合物a数量与质量的需求;实现质量分析人员对产品中该杂质残留量准确定量研究,从而确定合理的内控标准及货架期标准,确保产品安全有效;
[0038]
④
有利于降低雷替曲塞质量控制成本和检测成本;
[0039]
⑤
为叶酸类似物、抗代谢类靶点药物等新药的药物设计和药物合成提供借鉴。
4、附图说明
[0040]
图1为实施例2制备得到的化合物a的质谱图。
[0041]
图2为实施例2制备得到的化合物a高效液相纯度图谱。
[0042]
图3为实施例2制备得到的化合物a高效液相纯度图谱。
[0043]
图4为实施例5制备得到的化合物d质谱图。
[0044]
图5为实施例5制备得到的化合物d核测共振氢谱图。
[0045]
图6为实施例7市售注射用雷替曲塞(批号fh6736)高效液相色谱谱图。
5、具体实施方式
[0046]
下面详细描述本发明的具体实施例,需要说明的是下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
[0047]
实施例1中间体ⅱ的制备
[0048][0049]
500毫克化合物d,1.30克中间体ⅰ和360毫克2,6-二甲基吡啶加入到13毫升dmf中, 40℃搅拌反应40小时。tlc监控反应结束后降温,向体系中加入50毫升水和50毫升乙酸乙酯萃取分层,有机相用饱和食盐水洗涤,无水硫酸镁干燥后减压浓缩得粗品。粗品经硅胶色谱柱分离纯化(二氯甲烷∶甲醇=100:1至30:1梯度洗脱),得690毫克黄棕色固体,收率: 53.8%。
[0050]
实施例2化合物a的制备
[0051][0052]
260毫克中间体ⅱ加入到2.5毫升水中,加入60毫克氢氧化钠,室温搅拌反应4小时,tlc检测至反应完全。混合物中加入10毫升乙酸乙酯,萃取分层,水相用1摩尔每升hcl 溶液调节ph为4,有固体析出,继续搅拌30分钟,过滤,滤饼用3毫升水洗涤,减压干燥,得140毫克黄色固体,收率62%,液相纯度:97.2%。
[0053]
ms:检测值m/z,743[m+h]+,理论值m+h=743.18。
[0054]
1h-nmr(600mhz,dmso-d6):δ1.82~1.89(2h,overlap),1.98~2.04(2h,overlap), 2.30(4h,m),3.02(3h,s),3.16(3h,s),4.25~4.30(2h,overlap),4.48(2h,s), 4.65(2h,s),5.96(1h,d,j=4.2hz),5.99(1h,d,j=4.2hz),7.53(1h,d,j=4.2hz), 7.56
(1h,d,j=4.2hz),7.56(1h,d,j=8.4hz),7.65(1h,dd,j=8.4,1.2hz),7.96(1h, d,j=1.2hz),8.09(1h,d,j=6.0hz),8.10(1h,d,j=7.2hz),12.35(1h,brs),12.43 (4h,brs)。
[0055]
实施例3化合物a的制备
[0056]
300毫克中间体ⅱ加入到3毫升dmf中溶解,加入4m的氢氧化钾水溶液2毫升,室温搅拌反应4小时,tlc监控反应完全。加入10毫升二氯甲烷和5毫升水,萃取分层,有机相水洗后,合并水相。用盐酸调节水相ph为4,二氯甲烷萃取3次,每次5毫升。有机萃取液合并后减压蒸干,得320毫克粘稠剩余物。硅胶色谱柱层析分离纯化,二氯甲烷:甲醇=50:1 至8:1梯度洗脱,取目标物洗脱液减压蒸干得到淡黄色固体160毫克,收率:61.4%,液相纯度>98%。
[0057]
实施例4化合物d-2(2-溴甲基-6-甲基-3,4-二氢喹唑啉-4-酮)的制备
[0058]
8.2克氯乙腈溶于100毫升甲醇中,冰水浴冷却下加入1.1克甲醇钠,缓慢升温至16℃搅拌反应1小时。向体系中加入15.0克2-氨基-5-甲基苯甲酸后体系升温至65℃,保温搅拌2 小时。反应结束后降温析晶,滤饼用30毫升甲醇淋洗,得16.8克浅灰色固体。将固体投入 200毫升thf中,加入68.2克溴化锂,加热回流反应24小时。反应结束后冰水浴降温析晶,过滤得到滤饼用甲醇打浆两次得到浅黄色固体7.4克,收率29.3%。
[0059]
实施例5化合物d(2,6-二溴甲基-3,4-二氢喹唑啉-4-酮)的制备
[0060]
1.2克化合物d-2溶解于25毫升三氯甲烷中,加入160毫克aibn和1.3克nbs,升温至60℃搅拌反应15小时。反应体系冰水浴降温析晶,过滤,滤饼用10毫升氯仿淋洗,得浅黄色固体。所得固体用10毫升甲醇打浆洗涤得1.2克浅黄色固体,收率:76.4%。
[0061]
ms:检测值:333.0[m+h]+,354.9[m+na]+;理论值:m+h=332.85;m+na=354.83。
[0062]
1h-nmr(600mhz,dmso-d6):δ4.41(2h,s),4.89(2h,s),7.67(1h,d,j=8.4hz), 7.90(1h,d,j=8.4hz),8.20(1h,s),12.63(1h,s)。
[0063]
实施例6雷替曲塞有关物质化合物a的含量研究
[0064]
50.0克化合物c投入400毫升dmf中搅拌溶解,加入适量2,6-二溴甲基-3,4-二氢喹唑啉
ꢀ‑
4-酮(化合物d)作为加标验证对照。经液相检测化合物d在化合物c的溶液中含量为0.998%。按照专利cn106957296a报道的合成方法制备雷替曲塞,并对过程中产生的杂质进行跟踪检测,以确定化合物a的产生风险。
[0065]
表1雷替曲塞工艺有关物质
[0066]
有关物质化合物c中检出量雷替曲塞中间体中检出量雷替曲塞中检出量化合物d0.998%ndnd中间体
ⅱ‑‑
0.239nd化合物a
‑‑‑‑
0.022
[0067]
试验中涉及化合物列表如下:
[0068][0069][0070]
根据加标试验结果,化合物c中化合物d的含量控制在1.0%以下,化合物a在雷替曲塞产品中含量超过常规杂质限定量的风险较小。
[0071]
如果化合物c中化合物d的含量超过1.0%,可以采用如下方法处理化合物c以达到杂质控制目的:29.5克化合物c投入240毫升乙酸乙酯中,20-40℃搅拌浆洗4小时。过滤,滤饼用30毫升乙酸乙酯洗涤后室温减压干燥至恒重,得到27.3克类白色固体,收率92.5%。
[0072]
实施例7市售注射用雷替曲塞(批号fh6736)高效液相色谱检测
[0073]
照高效液相色谱法(中国药典2020年版四部通则0512)测定。
[0074]
溶剂乙腈-水(30︰70)。
[0075]
供试品溶液临用新制。取本品适量,加溶剂适量,超声使溶解,并用溶剂稀释制成每 1ml中约含0.4mg的溶液。
[0076]
系统适用性试验溶液取化合物a对照品适量,加溶剂使溶解并稀释制成每1ml中分别约含4μg的溶液。
[0077]
对照溶液精密量取供试品溶液适量,用溶剂定量稀释制成每1ml中约含0.4μg的溶
液。
[0078]
灵敏度溶液精密量取对照溶液5ml,置10ml量瓶中,用溶剂稀释至刻度,摇匀。
[0079]
色谱条件用十八烷基硅烷键合硅胶为填充剂(inertsil ods-3,4.6mm
×
150mm,3μm或效能相当的色谱柱);以10mmol/l磷酸二氢钾溶液为流动相a,以乙腈为流动相b,按下表进行梯度洗脱;流速为每分钟1.0ml;柱温为35℃;检测波长为228nm;进样体积10μl。
[0080][0081]
测定法精密量取供试品溶液与对照溶液,分别注入液相色谱仪,记录色谱图。高效液相色谱图谱见附图6。
[0082]
结果雷替曲塞出峰位置9.681分钟,化合物a出峰位置13.825,化合物a相对保留时间rrt=1.44。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1