CyclotheonellazoleA结构类似物及其合成方法和应用与流程
本发明涉及药物,尤其涉及一种cyclotheonellazole a结构类似物及其合成方法和应用。
背景技术:
1、由严重急性呼吸综合征冠状病毒2(sars-cov-2)引起的covid-19已成为严重威胁人类生命健康的全球流行病。研究表明,冠状病毒引起的急性肺损伤/急性呼吸窘迫综合征(ali/ards)是导致危重患者预后不良和生存率低的主要原因。目前,疫苗被认为是阻击sars-cov-2的最有效的方法。然而,随着新冠病毒变种的出现,导致病毒对疫苗诱导免疫的敏感性降低,因此,开发治疗sars-cov-2的药物是目前的重中之重。
2、sars-cov-2病毒感染宿主细胞,其路径具体为:sars-cov-2病毒与宿主细胞的受体蛋白血管紧张素转化酶2结合,而后在跨膜丝氨酸蛋白酶2(tmprss2)等作用下,病毒与宿主细胞膜发生膜融合,病毒基因组被释放到宿主细胞的细胞质中,进一步在主蛋白酶(mainprotease,mpro)等作用下,进行转录复制,完成子代病毒的组装,释放至胞外。
3、中性粒细胞弹性蛋白酶(neutrophilelastase,ne)是一种广泛表达于胰腺和中性粒细胞的丝氨酸蛋白酶。正常生理条件下,ne能够清除细菌、受损组织和促进组织再生,而在病理条件下,过度表达的弹性蛋白酶会损伤血管,从而导致炎症和病毒或细菌感染。研究表明,在covid-19患者中,中性粒细胞释放的弹性蛋白酶能够激活上皮na+转运体,导致高血压和肺气道脱水,从而使得黏液纤毛清除效率降低、肺透过屏障功能障碍以及促炎细胞因子释放,最终导致covid-19患者出现严重的ali/ards。因此,mpro和ne均被认为是治疗sars-cov-2的潜在靶点。
4、cyclotheonellazole a(简称为ca)是由以色列特拉维夫大学carmeli等人从海绵中分离得到的大环多肽类天然产物。从结构上看,ca由8个氨基酸构成,其中6个属于非蛋白原氨基酸,且含有七个手性中心。
5、
6、天然产物作为动植物、微生物和海洋生物的次级代谢产物,具有结构及生物活性的多样性和独特性。尽管该天然产物活性极高,但直接成药的可能性并不大。对其进行系统的构效关系研究,发现结构更简单、选择性高且治疗窗口良好的结构类似物是非常有必要的。
7、基于ca结构的复杂性,其具有众多的立体异构体,不同的立体异构体之间的生物活性往往千差万别,研究发现更多的天然产物立体异构体,并确证他们的蛋白酶抑制活性,对于构效关系的研究以及酶抑制剂候选药物的储备具有非常重要的价值。
技术实现思路
1、为了解决现有技术的问题,本发明实施例提供了一种cyclotheonellazole a结构类似物及其合成方法和应用。所述技术方案如下:
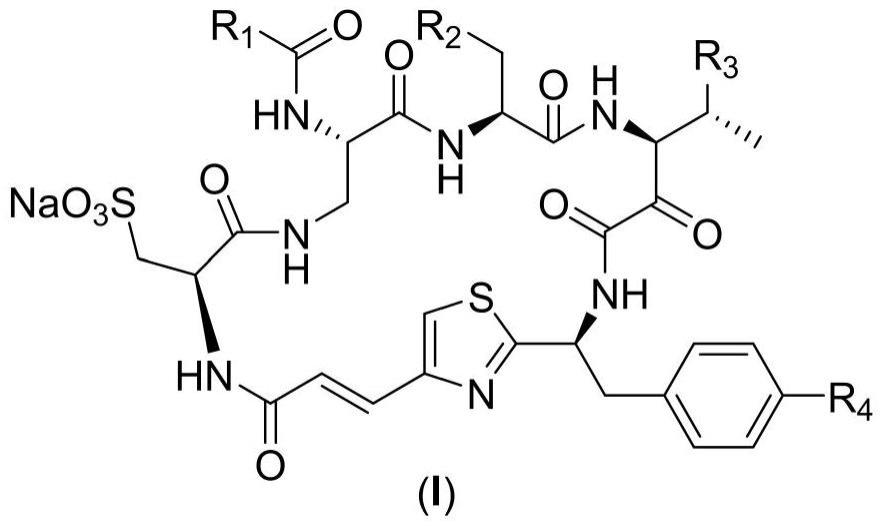
2、第一方面,提供一种具有通式(i)所示结构的化合物及其药学上可接受盐,所述通式(i)的结构如下:
3、
4、其中,r1、r2、r3、r4独立的选自h、c1-c6的烷基、c1-c6的烷氧基、卤素、羟基、氨基、硝基、氰基或巯基。
5、进一步的,所述r1为h,所述r2为c1-c3的烷基,所述r3为c1-c3的烷基,所述r4为羟基。
6、进一步的,所述化合物的结构为:
7、
8、第二方面,提供一种cyclotheonellazole a结构类似物的合成方法,所述方法包括:
9、
10、二乙胺脱掉化合物2的fmoc保护基,然后与甲酸乙酯在封管80℃下反应得到甲酰胺;用氟化铵脱掉tbdps保护基;用mscl活化羟基;用硫代乙酸钾发生sn2反应得到硫酯;用oxone氧化硫酯得到磺酸;接着用三乙胺在甲醇回流条件下脱掉特戊酰基得到仲醇;然后用ibx氧化仲醇得到α-酮酰胺;用二氧化硒氧化脱掉两个烯丙基得到磺酸,最后向磺酸化合物中加入饱和的氯化钠溶液形成磺酸钠盐,得到化合物1a;
11、其中,化合物2的结构如下:
12、
13、进一步的,所述化合物2的合成方法包括:
14、取化合物19和化合物20进行缩合反应得到化合物21,然后用tbscl保护化合物21的伯醇得到化合物22,用劳森试剂硫代化化合物22得到化合物23;用氟化铵脱掉化合物23的tbs保护基得到化合物24,用dast关环,dbu和三氯溴甲烷氧化,最后上烯丙基得到化合物25;用硼氢化锂还原化合物25的甲酯,用ibx氧化化合物25的伯醇,再通过wittig反应得到化合物26;用氢氧化钠水解化合物26的乙酯,再与三氯乙醇发生酯化反应得到化合物27;
15、
16、用三氟乙酸脱掉化合物27的boc保护基,然后与化合物18发生接肽反应得到化合物28;用三氟乙酸脱掉化合物28的boc保护基,然后与化合物11发生接肽反应得到化合物29;
17、
18、用锌粉在醋酸溶液中水解化合物29的三氯乙酯,接着用三氟乙酸脱掉boc保护基,最后用hatu在二氯甲烷溶液中发生分子内的关大环反应得到化合物2;
19、
20、其中,化合物11和化合物18的结构如下:
21、
22、进一步的,所述化合物18的合成方法包括:
23、以化合物12为起始原料,首先甲酯化,再用boc酸酐保护氨基,然后用硼氢化锂还原甲酯得到化合物13;接着用ibx氧化化合物13的伯醇得到醛,然后与丙酮氰醇反应,不经纯化直接用氯化氢的甲醇溶液回流水解后再用boc酸酐保护氨基得到化合物14和化合物14’两个非对映异构体;
24、
25、将化合物14与特戊酰氯反应得到化合物15,然后用三氟乙酸脱去化合物15的boc保护基,再与化合物16发生接肽反应得到化合物17;最后用碘化锂在吡啶回流条件下水解甲酯得到化合物18;
26、
27、其中,化合物16的结构如下:
28、
29、进一步的,所述化合物11的合成方法包括:
30、用三氟乙酸脱化合物8的boc保护基,然后与化合物9发生接肽反应得到化合物10;最后用锌粉在醋酸溶液中脱掉化合物10的pac保护基得到化合物11;
31、
32、其中,化合物9的结构如下:
33、
34、进一步的,所述化合物8的合成方法包括:
35、以化合物6为起始原料,首先用fmoc-osu保护氨基得到化合物7,再进行hoffman降解反应得到化合物3;然后用boc酸酐保护化合物3的氨基,用pacbr保护化合物3的羧基得到化合物8;
36、
37、第三方面,提供一种药物组合物,所述药物组合物包括上述第一方面任一项所述的化合物及其药学上可接受盐。
38、第四方面,提供一种上述第一方面任一项化合物及其药学上可接受盐在制备蛋白酶抑制药物中的应用。
39、本发明实施例提供的技术方案带来的有益效果是:本发明利用经典的逆合成分析,在cyclotheonellazole a全合成路线的基础上,有目的性地进行结构改造,天然产物的母核保持不变,而将左边的侧链替换为简单的甲酰胺得到结构类似物1a(tm),确证了cyclotheonellazole a结构类似物的极其优良的蛋白酶抑制活性,具有很强的医药工业的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!