吲唑并喹喔啉衍生物及其合成方法和应用
1.本发明涉及药物合成和医药技术领域,具体涉及一种吲唑并喹喔啉衍生物及其合成方法和应用。
背景技术:
2.癌症是起源于上皮组织的恶性肿瘤,是以细胞的快速增殖和转移为特点的疾病,其致死率高,是全球第二大死亡原因,严重威胁人类的健康,因此,医药领域一直在进行与癌症治疗有关的研究。
3.临床上治疗癌症的方式主要有:手术治疗、放射线治疗、化学治疗和免疫治疗等。其中,化学治疗是用可以抑制癌细胞生长或者杀死癌细胞的药物治疗癌症。目前,临床上用于治疗癌症疾病的传统药物有很多,但是大多具有容易产生耐药,特异性低,选择性差,毒副作用大等缺点。因此,有必要研发新的小分子抗肿瘤药物。
4.吲唑并环类化合物存在于许多天然产物、生物活性化合物和荧光探针中,例如mk-4827(抗癌)和pazopanib(酪氨酸激酶抑制剂)药物均属于吲唑并环类化合物。因其具有广泛的药理和生物活性而在医药和有机化学领域得到了广泛研究。研究者们试图合成更多新吲唑并环类化合物,以开发新型小分子抗肿瘤药物。
5.但是,已报道的吲唑并环类化合物的合成方法都存在一些缺陷,使更多新吲唑并环类化合物的合成和开发受到限制。例如,2h吲唑3位碳上的官能化是用到的比较多的方法,但是该方法需要使用重金属为催化剂,容易导致产物中贵金属超标,且原料官能化要求较高。此外,以3-氨基吲唑和异腈为原料可以得到吲唑并环类化合物,该方法具有较好的原子经济性,但底物扩展性较差,难以获得更多新型吲唑并环类化合物。
6.因此,有必要发展新型、高效合成吲唑并环类化合物的方法,以合成更多新型吲唑并环类化合物,以开发新型小分子抗肿瘤药物。
技术实现要素:
7.基于此,本发明提供了一种新型的高效合成吲唑并喹喔啉衍生物的方法,并利用该方法合成了一系列新的吲唑并喹喔啉衍生物,该类吲唑并喹喔啉衍生物对肝癌、乳腺癌、结肠癌等多种肿瘤有较强的抑制和治疗作用。
8.具体包括如下技术方案。
9.具有式(ⅰ)所示结构的吲唑并喹喔啉衍生物或者其药学上可接受的盐:
[0010][0011]
其中,r1选自:氢、c
1-c
12
烷基、c
6-c
10
芳基、5-10元杂芳基、卤素;
[0012]
r2选自:r5取代或者未取代的c
6-c
10
芳基、r5取代或者未取代的5-10元杂芳基、r6取代或者未取代的c
1-c
12
烷基;
[0013]
r3选自:氢、c
1-c
12
烷基、c
6-c
10
芳基、5-10元杂芳基、卤素;
[0014]
r4选自:r5取代或者未取代的c
6-c
10
芳基、r5取代或者未取代的5-10元杂芳基、r6取代或者未取代的c
1-c
12
烷基;
[0015]
r5选自:氢、c
1-c6烷基、c
6-c
10
芳基、5-6元杂芳基、卤素;
[0016]
r6选自:氢、c
1-c6烷基、c
6-c
10
芳基、5-6元杂芳基、卤素。
[0017]
在其中一些实施例中,r1选自:氢、c
1-c6烷基、卤素。
[0018]
在其中一些实施例中,r1选自:氢、甲基、乙基、正丙基、异丙基、氟、氯、溴。
[0019]
在其中一些实施例中,r2选自:r5取代或者未取代的苯基、r5取代或者未取代的噻吩基、苯基取代或者未取代的c
1-c6烷基;r5选自:氢、甲基、乙基、正丙基、异丙基、氟、氯、溴。
[0020]
在其中一些实施例中,r2选自:4-氯苯基、苯基、4-甲基苯基、噻吩基、苯乙基、苄基、苯丙基。
[0021]
在其中一些实施例中,r3选自:氢、c
1-c6烷基、卤素。
[0022]
在其中一些实施例中,r3选自:氢、甲基、乙基、正丙基、异丙基、氟、氯、溴。
[0023]
在其中一些实施例中,r4选自:c
4-c
10
烷基、r5取代或者未取代的苯基、苯基取代的c
1-c3烷基;r5选自:氢、甲基、乙基、正丙基、异丙基、氟、氯、溴。
[0024]
在其中一些实施例中,r4选自:c7烷基、c8烷基、c9烷基、c
10
烷基、r5取代或者未取代的苯基;r5选自:氢、甲基、乙基、正丙基、异丙基。
[0025]
在其中一些实施例中,r4选自:叔丁基、正丁基、2,6-二甲基苯基、1,1,3,3-四甲基丁基、苄基。
[0026]
一种上述的吲唑并喹喔啉衍生物的制备方法,包括如下步骤:
[0027]
(1)化合物(a)、化合物(b)、化合物(c)和化合物(d)在有机溶剂中反应,得化合物(e);
[0028]
(2)将化合物(e)溶于有机溶剂中,在催化剂和碱的作用下,于微波反应器中反应,即得式(i)所示的吲唑并喹喔啉衍生物;
[0029]
其反应式如下:
[0030][0031]
其中,x为卤素,r1、r2、r3、r4如前所述。
[0032]
在其中一些实施例中,所述化合物(a)、化合物(b)、化合物(c)和化合物(d)的摩尔比为1:1-1.2:1-1.2:1-1.2。
[0033]
在其中一些实施例中,步骤(1)所述有机溶剂为甲醇。
[0034]
在其中一些实施例中,步骤(1)所述反应温度为15℃-40℃。
[0035]
在其中一些实施例中,步骤(2)所述有机溶剂为n,n-二甲基甲酰胺。
[0036]
在其中一些实施例中,步骤(2)所述催化剂为铜催化剂和n,n-四甲基乙二胺。
[0037]
在其中一些实施例中,所述铜催化剂和n,n-四甲基乙二胺的摩尔比为1:1.5-2.5。
[0038]
在其中一些实施例中,所述铜催化剂选自碘化亚铜、溴化亚铜、氧化亚铜、三氟甲烷磺酸铜。
[0039]
在其中一些实施例中,步骤(2)所述催化剂为摩尔比为1:1.5-2.5的碘化亚铜和n,n-四甲基乙二胺。
[0040]
在其中一些实施例中,步骤(2)所述碱选自碳酸钾、1,8-二氮杂双环[5.4.0]十一碳-7-烯、4-二甲氨基吡啶、叔丁醇钾、碳酸铯。
[0041]
在其中一些实施例中,步骤(2)所述碱为碳酸钾。
[0042]
在其中一些实施例中,步骤(2)所述化合物(e)和碱的摩尔比为1:1.5-2.5。
[0043]
在其中一些实施例中,步骤(2)所述反应的温度为140℃-160℃,反应的时间为20min-40min。
[0044]
在其中一些实施例中,步骤(2)所述反应的温度为145℃-155℃,反应的时间为25min-35min。
[0045]
本发明还提供了上述的吲唑并喹喔啉衍生物或者其药学上可接受的盐的应用,包括如下技术方案。
[0046]
上述的吲唑并喹喔啉衍生物或者其药学上可接受的盐在制备预防和/或治疗肿瘤的药物中的应用。
[0047]
在其中一些实施例中,所述肿瘤为:肝癌、乳腺癌、结肠癌。
[0048]
本发明还提供了一种预防和/或治疗肿瘤的药用组合物,包括如下技术方案。
[0049]
一种预防和/或治疗肿瘤的药用组合物,由活性成分和药学上可接受的辅料制备得到,所述活性成分包括上述的吲唑并喹喔啉衍生物或者其药学上可接受的盐。
[0050]
本发明以吲唑-3-羧酸、苯胺、醛和异腈为原料,经过ugi反应和碱性条件下的一锅
法关环反应制备得到了一系列新的吲唑并喹喔啉衍生物,首次合成出了具有抗肿瘤活性的吲唑并喹喔啉衍生物,对其进行体外肿瘤细胞抑制活性的测试结果显示这类化合物对肝癌、乳腺癌和结肠癌等肿瘤细胞有较强的抑制作用,具有较好的抗肿瘤活性,可用于制备抗肿瘤药物。
[0051]
本发明的吲唑并喹喔啉衍生物的合成方法具有操作工艺简单、合成路线短,成本低、收率高、底物适用广等优点。
具体实施方式
[0052]
本发明下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所用到的各种常用化学试剂,均为市售产品。
[0053]
除非另有定义,本发明所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不用于限制本发明。
[0054]
本发明的术语“包括”和“具有”以及它们任何变形,意图在于覆盖不排他的包含。例如包含了一系列步骤的过程、方法、装置、产品或设备没有限定于已列出的步骤或模块,而是可选地还包括没有列出的步骤,或可选地还包括对于这些过程、方法、产品或设备固有的其它步骤。
[0055]
在本发明中提及的“多个”是指两个或两个以上。“和/或”,描述关联对象的关联关系,表示可以存在三种关系,例如,a和/或b,可以表示:单独存在a,同时存在a和b,单独存在b这三种情况。字符“/”一般表示前后关联对象是一种“或”的关系。
[0056]
本发明所述化合物中,当任何变量在任何组分中出现超过一次,则其每次出现的定义独立于其它每次出现的定义。同样,允许取代基及变量的组合,只要这种组合使化合物稳定。自取代基划入环系统的线表示所指的键可连接到任何能取代的环原子上。如果环系统为多环,其意味着这种键仅连接到邻近环的任何适当的碳原子上。要理解本领域普通技术人员可选择本发明化合物的取代基及取代型式而提供化学上稳定的并可通过本领域技术和下列提出的方法自可容易获得的原料容易的合成的化合物。如果取代基自身被超过一个基团取代,应理解这些基团可在相同碳原子上或不同碳原子上,只要使结构稳定。
[0057]
本发明所用术语“烷基”意指包括具有特定碳原子数目的支链的和直链的饱和脂肪烃基。例如,“c
1-c6烷基”中“c
1-c
6”的定义包括以直链或支链排列的具有1、2、3、4、5或6个碳原子的基团。例如,“c
1-c6烷基”具体包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、异丁基、戊基、己基。
[0058]
本发明所用术语“杂芳基”指含有1个或多个选自o、n或s的杂原子的芳香环,本发明范围内的杂芳基包括但不限于:喹啉基、吡唑基、吡咯基、噻吩基、呋喃基、吡啶基、嘧啶基、吡嗪基、三氮唑基、咪唑基、恶唑基、异恶唑基、哒嗪基、苯并呋喃基、苯并噻吩基、苯并恶唑、吲哚基等;“杂芳基”也理解为包括任何含有氮的杂芳基的n-氧化物衍生物。杂芳基的连接可以通过碳原子或通过杂原子实现。
[0059]
本文所用术语“取代的”是指用指定取代基的基团置换特定结构中的氢基。
[0060]
正如本领域技术人员所理解的,本文中所用“卤素”(“halo”)或“卤”意指氯、氟、溴和碘。
[0061]
在一个实施方案中,本发明提供了一种利用具有式(ⅰ)所示结构的化合物及其药学上可接受的盐治疗人或其它哺乳动物的肿瘤疾病。
[0062]
在一个实施方案中,本技术的化合物及其药学上可接受的盐可以用于治疗或控制肝癌、乳腺癌、结肠癌等肿瘤。
[0063]
本发明还提供了一种药物组合物,它包含安全有效量范围内的活性成分,以及药学上可接受的载体或者辅料。
[0064]
本发明所述的“活性成分”是指本发明所述的式(ⅰ)化合物或者其药学上可接受的盐。
[0065]
本发明所述的“活性成分”和药物组合物可用于制备预防和/或治疗肿瘤的药物。
[0066]“安全有效量”指的是:活性成分的量足以明显改善病情,而不至于产生严重的副作用。
[0067]“药学上可接受的载体或者辅料”指的是:一种或多种相容性固体或液体填料或凝胶物质,它们适合于人使用,而且必须有足够的纯度和足够低的毒性。
[0068]“相容性”在此指的是组合物中各组分能和本发明的活性成分以及它们之间相互掺和,而不明显降低活性成分的药效。
[0069]
药学上可以接受的载体或者辅料部分例子有纤维素及其衍生物(如羧甲基纤维素钠、乙基纤维素钠、纤维素乙酸酯等)、明胶、滑石、固体润滑剂(如硬脂酸、硬脂酸镁)、硫酸钙、植物油(如豆油、芝麻油、花生油、橄榄油等)、多元醇(如丙二醇、甘油、甘露醇、山梨醇等)、乳化剂润湿剂(如十二烷基硫酸钠)、着色剂、调味剂、稳定剂、抗氧化剂、防腐剂、无热原水等。
[0070]
在另一优选例中,本发明式(ⅰ)化合物可与大分子化合物或高分子通过非键合作用形成复合物。在另一优选例中,本发明式(ⅰ)化合物作为小分子还可通过化学键与大分子化合物或高分子相连接。所述大分子化合物可以是生物大分子如高聚糖、蛋白、核酸、多肽等。
[0071]
本发明的活性成分或药物组合物的施用方式没有特别限制,代表性的施用方式包括(但并不限于):口服、瘤内、直肠、肠胃外(静脉内、肌肉内或皮下)等。
[0072]
用于口服给药的固体剂型包括胶囊剂、片剂、丸剂、散剂和颗粒剂。
[0073]
在这些固体剂型中,活性成分与至少一种常规惰性赋形剂(或载体)混合,如柠檬酸钠或磷酸二钙,或与下述成分混合:
[0074]
(a)填料或增容剂,例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸;
[0075]
(b)粘合剂,例如,羟甲基纤维素、藻酸盐、明胶、聚乙烯基吡咯烷酮、蔗糖和阿拉伯胶;
[0076]
(c)保湿剂,例如,甘油;
[0077]
(d)崩解剂,例如,琼脂、碳酸钙、马铃薯淀粉或木薯淀粉、藻酸、某些复合硅酸盐、和碳酸钠;
[0078]
(e)缓溶剂,例如石蜡;
[0079]
(f)吸收加速剂,例如,季胺化合物;
[0080]
(g)润湿剂,例如鲸蜡醇和单硬脂酸甘油酯;
[0081]
(h)吸附剂,例如,高岭土;和
[0082]
(i)润滑剂,例如,滑石、硬脂酸钙、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠,或其混合物。胶囊剂、片剂和丸剂中,剂型也可包含缓冲剂。
[0083]
所述的固体剂型还可采用包衣和壳材制备,如肠衣和其它本领域公知的材料。它们可包含不透明剂,并且,这种组合物中活性成分的释放可以延迟的方式在消化道内的某一部分中释放。可采用的包埋组分的实例是聚合物质和蜡类物质。
[0084]
用于口服给药的液体剂型包括药学上可接受的乳液、溶液、悬浮液、糖浆或酊剂。除了活性成分外,液体剂型可包含本领域中常规采用的惰性稀释剂,如水或其它溶剂,增溶剂和乳化剂,例知,乙醇、异丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油,特别是棉籽油、花生油、玉米胚油、橄榄油、蓖麻油和芝麻油或这些物质的混合物等。除了这些惰性稀释剂外,组合物也可包含助剂,如润湿剂、乳化剂和悬浮剂、甜味剂、矫味剂和香料。
[0085]
除了活性成分外,悬浮液可包含悬浮剂,例如,乙氧基化异十八烷醇、聚氧乙烯山梨醇和脱水山梨醇酯、微晶纤维素、甲醇铝和琼脂或这些物质的混合物等。
[0086]
用于肠胃外注射的组合物可包含生理上可接受的无菌含水或无水溶液、分散液、悬浮液或乳液,和用于重新溶解成无菌的可注射溶液或分散液的无菌粉末。适宜的含水和非水载体、稀释剂、溶剂或赋形剂包括水、乙醇、多元醇及其适宜的混合物。
[0087]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如sambrook等人,分子克隆:实验室手册(newyork:cold spring harbor laboratory press,1989)中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
[0088]
除非另行定义,文中所使用的所有专业与科学用语与本领域技术人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
[0089]
下列实施例中所用试剂均可购买得到。
[0090]
以下实施例中所述室温是指25
±
5℃的室内温度。
[0091]
以下实施例中所用微波反应器为拜泰齐贸易(上海)有限公司的biotage initiator+微波合成仪。
[0092]
实施例2-14中的吲唑并喹喔啉衍生物的具体合成路线如下:
[0093][0094]
其中,x为卤素;
[0095]
r1选自:氢、c
1-c
12
烷基、c
6-c
10
芳基、5-10元杂芳基、卤素;
[0096]
r2选自:r5取代或者未取代的c
6-c
10
芳基、r5取代或者未取代的5-10元杂芳基、r6取代或者未取代的c
1-c
12
烷基;
[0097]
r3选自:氢、c
1-c
12
烷基、c
6-c
10
芳基、5-10元杂芳基、卤素;
[0098]
r4选自:r5取代或者未取代的c
6-c
10
芳基、r5取代或者未取代的5-10元杂芳基、r6取代或者未取代的c
1-c
12
烷基;
[0099]
r5选自:氢、c
1-c6烷基、c
6-c
10
芳基、5-6元杂芳基、卤素;
[0100]
r6选自:氢、c
1-c6烷基、c
6-c
10
芳基、5-6元杂芳基、卤素。
[0101]
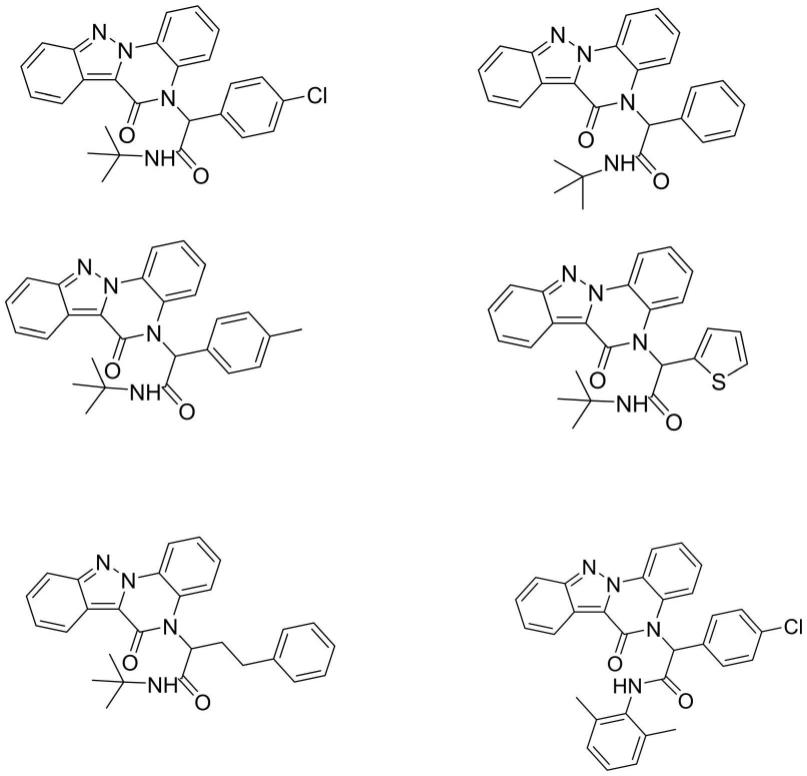
实施例1n-叔丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺的合成:
[0102][0103]
步骤(1):在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,残留物用硅胶柱分离得到化合物5a,产率85%。
[0104]1h nmr(400mhz,cdcl3)δ10.59(s,1h),8.13(d,j=8.2hz,1h),7.89(d,j=7.6hz,1h),7.30(m,4h),7.19
–
7.07(m,5h),6.89(t,j=7.3hz,1h),6.40(s,1h),6.18(s,1h),1.37(s,9h).
13
c nmr(101mhz,cdcl3)δ168.88,164.26,139.90,139.27,134.44,133.15,133.03,132.39,132.11,131.70,129.12,128.57,128.11,127.56,126.90,125.59,123.46,122.38,122.23,109.86,66.14,51.80,28.66.hrms(esi)m/z calcd for c
26h25
brcln4o
2+
(m+h)
+
539.0844,found 539.0838.
[0105]
步骤(2):将化合物5a用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碱(2.0毫摩尔),催化剂1(0.1毫摩尔),催化剂2(0.2毫摩尔),在微波反应器中反应。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺(化合物6a)。
[0106]1h nmr(400mhz,cdcl3)δ8.68(dd,j=7.8,1.8hz,1h),8.38(d,j=8.4hz,1h),8.00(d,j=8.7hz,1h),7.61
–
7.49(m,2h),7.47
–
7.36(m,3h),7.32(q,j=8.9hz,4h),6.98(s,1h),5.99(s,1h),1.33(s,9h).
13
c nmr(101mhz,cdcl3)δ166.50,155.64,149.05,134.04,132.56,129.27,128.94(s),128.30,128.20,125.23,124.51,122.70,121.85,121.00,118.47,117.84,117.73,59.10,52.24,28.56.hrms(esi)m/z calcd for c
26h24
cln4o
2+
(m+h)
+
459.1582,found 459.1580.
[0107]
其中,步骤(2)所述碱、催化剂1和催化剂2以及反应温度、时间和收率如表1所示。
[0108]
表1
[0109]
序号碱和催化剂反应温度(℃)时间(min)6a收率(%)1k2co3mw 12010mintrace2k2co3mw 15010min253k2co3mw 15030min55
4teamw 15030mintrace5dbumw 15030min156dmapmw 15030min177kotbumw 15030min318cs2co3mw 15030min439k2co
3 cui tmedamw 15030min9510k2co
3 cubr tmedamw 15030min6511k2co
3 cu2o tmedamw 15030min5312k2co
3 cu(otf)
2 tmedamw 15030min49
[0110]
实施例2n-叔丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺的合成:
[0111][0112]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(tmeda、0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺,产率73%。
[0113]1h nmr(400mhz,cdcl3)δ8.68(dd,j=7.8,1.8hz,1h),8.38(d,j=8.4hz,1h),8.00(d,j=8.7hz,1h),7.61
–
7.49(m,2h),7.47
–
7.36(m,3h),7.32(q,j=8.9hz,4h),6.98(s,1h),5.99(s,1h),1.33(s,9h).
13
c nmr(101mhz,cdcl3)δ166.50,155.64,149.05,134.04,132.56,129.27,128.94(s),128.30,128.20,125.23,124.51,122.70,121.85,121.00,118.47,117.84,117.73,59.10,52.24,28.56.hrms(esi)m/z calcd for c
26h24
cln4o
2+
(m+h)
+
459.1582,found 459.1580.
[0114]
实施例3n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺的合成:
[0115]
在5毫升微波反应管中,先将苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入
该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺,产率67%。
[0116]1h nmr(400mhz,cdcl3)δ8.66(dd,j=7.8,1.9hz,1h),8.37(d,j=8.4hz,1h),7.98(d,j=8.7hz,1h),7.60
–
7.51(m,2h),7.46
–
7.27(m,8h),6.94(s,1h),6.02(s,1h),1.35(s,9h).
13
c nmr(101mhz,cdcl3)δ166.70,155.70,149.15,134.16,129.87,128.97,128.27,128.09,127.83,125.17,125.09,124.28,123.04,121.94,121.35118.35,117.79,117.65,60.54,52.12,28.57.hrms(esi)m/z calcd for c
26h25
n4o
2+
(m+h)
+
425.1972,found 425.1974.
[0117]
实施例4 n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-对甲苯基乙酰胺的合成:
[0118][0119]
在5毫升微波反应管中,先将对甲基苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-对甲苯基乙酰胺,产率68%。
[0120]1h nmr(400mhz,cdcl3)δ8.66(d,j=9.7hz,1h),8.39(d,j=8.4hz,1h),8.03
–
7.93(m,1h),7.60
–
7.51(m,2h),7.45
–
7.33(m,3h),7.27
–
7.19(m,3h),7.13(d,j=7.1hz,1h),6.85(s,1h),5.98(s,1h),2.31(s,3h),1.35(s,9h).
13
c nmr(101mhz,cdcl3)δ166.74,155.69,149.16,138.87,134.16,130.02,129.15,128.88,128.44,128.26,128.09,125.17,125.05,124.86,124.22,123.13,121.94,121.42,118.26,117.77,117.62,60.81,52.08,28.58,21.58.hrms(esi)m/z calcd for c
27h27
n4o
2+
(m+h)
+
439.2129,found 439.2129.
[0121]
实施例5 n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-(噻吩-2-基)乙酰胺的合成:
[0122][0123]
在5毫升微波反应管中,先将噻吩-2-甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-(噻吩-2-基)乙酰胺,产率53%。
[0124]1h nmr(400mhz,cdcl3)δ8.69
–
8.62(m,1h),8.39(d,j=8.4hz,1h),7.97(d,j=8.7hz,1h),7.56(ddd,j=8.8,4.9,1.6hz,2h),7.46
–
7.37(m,3h),7.32(dd,j=5.1,1.1hz,2h),7.09(d,j=3.0hz,1h),6.94(dd,j=5.1,3.7hz,1h),5.95(s,1h),5.29(s,1h),1.33(s,9h).
13
c nmr(101mhz,cdcl3)δ166.23,155.20,149.14,135.75,128.34,128.21,128.09,127.38,126.49,125.26,124.44,122.00,121.28,117.85,53.45,52.25,28.52.hrms(esi)m/z calcd for c
24h23
n4o2s
+
(m+h)
+
431.1536,found 431.1539.
[0125]
实施例6 n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-4-苯基丁酰胺的合成:
[0126][0127]
在5毫升微波反应管中,先将苯丙醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-4-苯基丁酰胺,产率62%。
[0128]1h nmr(400mhz,cdcl3)δ8.58(dd,j=8.1,1.3hz,1h),8.30(d,j=8.4hz,1h),7.95(d,j=8.7hz,1h),7.65(d,j=7.9hz,1h),7.58
–
7.52(m,1h),7.51
–
7.46(m,1h),7.45
–
7.36(m,2h),6.94(s,5h),6.27(s,1h),5.87(s,1h),2.80(ddd,j=21.6,13.4,7.2hz,2h),2.48(dd,j=11.1,6.1hz,2h),1.28(s,9h).
13
c nmr(101mhz,cdcl3)δ168.93,155.94,149.01,140.11,128.29,128.02,127.99,125.83,125.22,125.16,124.42,121.79,121.04,117.94,
117.80,55.21,51.82,33.01,28.55.hrms(esi)m/z calcd for c
28h29
n4o
2+
(m+h)
+
453.2285,found 453.2286.
[0129]
实施例7 2-(4-氯苯基)-n-(2,6-二甲基苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺的合成:
[0130][0131]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和2,6-二甲基苯基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物2-(4-氯苯基)-n-(2,6-二甲基苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺,产率56%。
[0132]1h nmr(400mhz,cdcl3)δ8.67(d,j=8.0hz,1h),8.33(d,j=8.3hz,1h),7.98(d,j=8.7hz,1h),7.68
–
7.52(m,3h),7.49(d,j=8.4hz,2h),7.40(ddd,j=20.5,10.5,7.2hz,5h),7.22(s,1h),7.04(dt,j=17.4,6.6hz,3h),2.13(s,6h).
13
c nmr(101mhz,cdcl3)δ165.72,155.76,149.19,135.16,134.41,133.05,132.14,129.31,129.21,128.47,128.34,127.64,125.46,125.31,124.78,122.69,121.99,121.13,118.24,117.96,117.90,58.84,18.47.hrms(esi)m/z calcd for c
30h24
cln4o
2+
(m+h)
+
507.1582,found 507.1572.
[0133]
实施例8 2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-n-(2,4,4-三甲基戊烷-2-基)乙酰胺的合成:
[0134][0135]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和1,1,3,3-四甲基丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次
加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-n-(2,4,4-三甲基戊烷-2-基)乙酰胺,产率66%。
[0136]1h nmr(400mhz,cdcl3)δ8.65(d,j=7.6hz,1h),8.34(d,j=8.4hz,1h),7.97(d,j=8.7hz,1h),7.55(dd,j=15.5,7.6hz,2h),7.36(ddd,j=26.0,16.2,8.0hz,7h),6.97(s,1h),6.11(s,1h),1.64(q,j=15.0hz,2h),1.39(d,j=13.2hz,6h),0.80(s,9h).
13
c nmr(101mhz,cdcl3)δ165.98,155.55,149.14,134.04,132.33,129.36,128.91,128.36,128.29,125.28,125.16,124.56,122.71,121.93,121.19,118.37,117.89,117.77,59.13,56.19,52.45,31.50,31.20,28.85,28.32.hrms(esi)m/z calcd for c
30h32
cln4o
2+
(m+h)
+
515.2208,found 515.2210.
[0137]
实施例9 n-苄基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺的合成:
[0138][0139]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和苄基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机项使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-苄基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺,产率76%。
[0140]1h nmr(400mhz,cdcl3)δ8.57(dd,j=7.9,1.8hz,1h),8.10(d,j=8.4hz,1h),7.92(d,j=8.7hz,1h),7.55
–
7.45(m,2h),7.41
–
7.31(m,4h),7.31
–
7.26(m,3h),7.21
–
7.11(m,5h),7.04(s,1h),6.84(t,j=5.3hz,1h),4.55
–
4.42(m,2h).
13
c nmr(101mhz,cdcl3)δ167.28,155.65,149.01,137.50,134.25,132.08,129.34,129.18,129.04,128.61,128.33,127.72,127.53,125.32,125.22,124.58,122.59,121.79,120.88,118.13,117.84,58.38,44.08.hrms(esi)m/z calcd for c
29h22
cln4o
2+
(m+h)
+
493.1426,found 493.1419.
[0141]
实施例10 n-丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺的合成:
[0142][0143]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和正丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-丁基-2-(4-氯苯基)-2-(6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)乙酰胺,产率57%。
[0144]1h nmr(400mhz,cdcl3)δ8.57(d,j=7.2hz,1h),8.16(d,j=8.3hz,1h),7.92(d,j=8.7hz,1h),7.51(dd,j=14.9,7.5hz,2h),7.40
–
7.26(m,7h),7.01(s,1h),6.57(s,1h),3.23(dd,j=13.3,6.6hz,2h),1.46
–
1.35(m,2h),1.28
–
1.13(m,2h),0.80(t,j=7.3hz,3h).
13
c nmr(101mhz,cdcl3)δ167.23,155.63,148.99,134.13,132.30,129.33,129.26,128.97,128.32,125.25,125.16,124.53,122.62,121.73,120.88,118.19,117.84,117.74,58.45,39.88,31.27,19.99,13.66.hrms(esi)m/z calcd for c
26h24
cln4o
2+
(m+h)
+
459.1582,found 459.1571.
[0145]
实施例11 n-叔丁基-2-(2-氯-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-(4-氯苯基)乙酰胺的合成:
[0146][0147]
在5毫升微波反应管中,先将对氯苯甲醛(1.0毫摩尔)和2-溴-4-氯苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(2-氯-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-(4-氯苯基)乙酰胺,产率
65%。
[0148]1h nmr(400mhz,cdcl3)δ8.63(d,j=2.1hz,1h),8.28(d,j=8.4hz,1h),7.95(d,j=8.7hz,1h),7.59
–
7.53(m,1h),7.50(d,j=9.1hz,1h),7.44
–
7.38(m,1h),7.34
–
7.25(m,6h),7.02(s,1h),6.15(s,1h),1.34(s,9h).
13
c nmr(101mhz,cdcl3)δ166.21,155.40,149.31,134.25,132.27,130.30,129.08,128.71,128.12,127.80,125.78,125.64,122.76,122.01,121.11,120.06,117.97,117.57,58.86,52.37,28.59.hrms(esi)m/z calcd for c
26h23
cl2n4o
2+
(m+h)
+
493.1193,found 493.1186.
[0149]
实施例12 n-叔丁基-2-(2-甲基-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺的合成:
[0150][0151]
在5毫升微波反应管中,先将苯甲醛(1.0毫摩尔)和2-溴4-甲基苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(2-甲基-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺,产率58%。
[0152]1h nmr(400mhz,cdcl3)δ8.47(d,j=1.0hz,1h),8.36(dd,j=8.4,0.9hz,1h),7.97(d,j=8.7hz,1h),7.58
–
7.50(m,1h),7.46
–
7.38(m,4h),7.38
–
7.27(m,3h),7.15(dd,j=8.7,1.5hz,1h),6.97(s,1h),6.06(s,1h),2.47(s,3h),1.34(s,9h).
13
c nmr(101mhz,cdcl3)δ166.78,155.60,149.10,134.61,134.25,129.13,128.90,128.24,128.17,127.81,127.56,124.97,123.10,121.91,121.37,118.32,117.68,117.55,60.21,52.09,28.59,20.82.hrms(esi)m/z calcd for c
27h27
n4o
2+
(m+h)
+
439.2129,found 439.2127.
[0153]
实施例13n-叔丁基-2-(8-甲基-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺的合成:
[0154][0155]
在5毫升微波反应管中,先将苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将5-甲基吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依
次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(8-甲基-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺,产率54%。
[0156]1h nmr(400mhz,cdcl3)δ8.62(dd,j=7.9,1.6hz,1h),8.14(s,1h),7.87(d,j=8.8hz,1h),7.55(dd,j=8.0,1.2hz,1h),7.37(ddt,j=18.2,10.2,7.2hz,8h),6.91(s,1h),6.02(s,1h),2.54(s,3h),1.35(s,9h).
13
c nmr(101mhz,cdcl3)δ166.79,155.85,148.07,135.20,134.22,131.12,129.75,128.93,128.22,127.83,125.32,124.22,122.33,122.25,119.64,118.29,117.48,117.43,60.51,52.08,28.58,22.00.hrms(esi)m/z calcd for c
27h27
n4o
2+
(m+h)
+
439.2129,found 439.2119.
[0157]
实施例14n-叔丁基-2-(9-氯-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺的合成:
[0158][0159]
在5毫升微波反应管中,先将苯甲醛(1.0毫摩尔)和邻溴苯胺(1.0毫摩尔)溶解在2.0毫升的甲醇中,然后再将6-氯吲唑-3-羧酸(1.0毫摩尔)和叔丁基异腈(1.0毫摩尔)依次加入该溶液中,反应液室温下搅拌12小时,然后使用薄层色谱检测,反应完成后,真空旋转蒸发除去溶剂,然后再用5.0毫升的n,n-二甲基甲酰胺(dmf)溶解,再依次加入碳酸钾(2.0毫摩尔),碘化亚铜(0.1毫摩尔),n,n-四甲基乙二胺(0.2毫摩尔),在微波反应器中150℃反应30分钟。冷却至室温后该溶液使用乙酸乙酯(15毫升)稀释,然后用饱和食盐水洗涤2次,每次20毫升。有机相使用无水硫酸钠干燥后,采用硅胶柱进行分离,得到目标化合物n-叔丁基-2-(9-氯-6-氧代吲唑并[2,3-a]喹喔啉-5(6h)-基)-2-苯基乙酰胺,产率46%。
[0160]1h nmr(400mhz,cdcl3)δ8.55(dd,j=6.2,3.5hz,1h),8.17(d,j=8.8hz,1h),7.90
–
7.84(m,1h),7.56(dd,j=6.2,3.4hz,1h),7.45
–
7.29(m,7h),7.25(dd,j=5.1,1.8hz,1h),6.92(s,1h),6.19(s,1h),1.34(s,9h).
13
c nmr(101mhz,cdcl3)δ166.59,155.42,149.22,134.17,129.94,129.06,128.42,128.32,127.88,126.28,124.92,124.39,123.33,122.39,120.13,118.40,117.66,116.85,60.78,52.17,28.58.hrms(esi)m/z calcd for c
26h24
cln4o
2+
(m+h)
+
459.1582,found 459.1583.
[0161]
实施例15抗肿瘤活性测试
[0162]
本实施例抗肿瘤测试所用的细胞为hep3b、mda-mb-453和hct116。
[0163]
细胞使用的培养液为含青霉素一链霉素溶液的胎牛血清的dmem细胞培养液,培养条件为37℃、含5%co2的恒温培养箱。具体步骤如下:
[0164]
(1)用血球计数板对细胞进行计数后,用dmem low glucose培养液将其稀释至5
×
104个/ml;
[0165]
(2)在96孔板的每个孔里加入100μl细胞悬液吹打混匀,培养箱37℃温育24h;
[0166]
(3)将待测试化合物稀释至浓度为10μm,按照该浓度依次加药,培养箱37℃温育48h;
[0167]
(4)加入浓度为5mg/ml的mtt,培养箱37℃温育4h;
[0168]
(5)加dmso将细胞溶解,酶标仪测490nm和630nm下的od值;(6)处理数据,根据od值计算抑制率,结果如表2所示。
[0169]
表2吲唑并喹喔啉衍生物的抗肿瘤活性结果(抑制率%)
[0170]
[0171][0172]
由测试结果可以看出,本发明的吲唑并喹喔啉衍生物可以有效抑制或杀灭肿瘤细胞,具有良好的抗肿瘤活性,可以用于制备治疗肝癌、乳腺癌和结肠癌等肿瘤的药物。
[0173]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对以下实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0174]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1