一类含有2-芳基-4-取代喹啉结构的化合物及其制备方法与应用
1.本发明属于医药技术领域,具体涉及一类含有2-芳基-4-取代喹啉结构的化合物的制备方法及其在制备抗肿瘤药物中的应用。
背景技术:
2.恶性肿瘤严重危害人类的健康。近年来,我国癌症发病率和死亡率逐年上升。据who全球癌症报告:2018年中国癌症发病人数为405.6万(约占全球癌症发病总人数的22%),其中癌症死亡人数为261.8万(约占全球癌症死亡总人数的27%)。目前临床上常用150多种抗肿瘤药物,但多数存在疗效有限、毒副作用大和耐药性严重等问题。因此,研发新结构类型、新作用机制、高效低毒的抗肿瘤创新药物一直是全球新药研发的热点。
3.微管是真核细胞中重要的组成部分,与其他蛋白共同组装成纺锤体、中心粒、鞭毛、神经管等多种结构。正常生理条件下,微管的聚合和解聚保持着动态平衡,因此细胞分裂高度可控,进展有序。这种不稳定的动力学特性使得微管在维持细胞形态、细胞迁移、细胞有丝分裂、胞内物质的输送及信号传导等细胞关键生物学过程中具有重要调节功能。微管在细胞分裂前期聚合成为纺锤体,而纺锤体在细胞有丝分裂过程中牵引染色体向两极移动进入至两个子细胞中,从而完成细胞增殖。微管的管壁由14条平行的原纤维包围而成,每根原纤维又是由αβ微管蛋白二聚体头尾依次相接而成。这种构造使微管形成了具有极性的α端和β端,由于微管在细胞有丝分裂过程中承担的重要作用,微管蛋白逐渐成为医药工作者研究与开发抗癌药物的重要靶点之一,而以微管蛋白为靶点的微管蛋白抑制剂也已成为临床证实有效的抗肿瘤药物。该类抑制剂的作用机制是靶向作用于血管内皮细胞的微管蛋白,影响微管的装配而干扰微管的正常结构与功能,阻止纺锤丝形成或使纺锤丝失去动力,姐妹染色单体在有丝分裂期失去分离的牵引力,无法正确双向取向进而干扰细胞的有丝分裂过程,使细胞有丝分裂中断,停滞于m期,从而导致肿瘤细胞发生凋亡,破坏内皮细胞骨架结构而造成肿瘤血管系统选择性闭塞和大规模损伤坏死,发挥抗肿瘤作用。
4.研究已发现微管蛋白存在8个抑制剂结合位点。其中紫杉醇位点、长春碱位点和秋水仙碱位点发现较早,作用机制已经相对成熟,并且紫杉醇和长春碱位点微管蛋白抑制剂类药物已在肿瘤临床治疗方面占据了重要地位,紫杉醇更是已成为肺癌、乳腺癌和卵巢癌的重要一线治疗药物。目前临床上广泛应用的微管蛋白抑制剂有紫杉醇、多西紫杉醇、长春花碱和长春瑞滨等。但这些药物存在难以耐受的副作用及用药后易产生耐药等问题,限制了微管蛋白抑制剂的临床使用。然而与紫杉醇位点和长春新碱位点的抑制剂相比,秋水仙碱结合位点的活性腔体积较小,便于研究;且秋水仙碱位点的抑制剂仍对紫杉醇类和长春碱类耐药的肿瘤株有效,并不易产生耐药性。因此,开发新型的具有更强活性、更低毒性、并且对多药耐药肿瘤株更有效的新型微管蛋白抑制剂具有很大的应用前景。
5.前期工作中,我们团队采用虚拟筛选的方法发现了一类2-芳基取代的喹啉结构衍生物为秋水仙碱结合位点的抑制剂,并表现中等、广谱的抗肿瘤活性(bioorg chem,2022,
118,105486)。在此基础上,本专利对前期发现的先导结构进行了结构改造,发现了一类全新结构的2-芳基-4-取代喹啉结构的化合物表现更为优异的抗肿瘤活性,酶实验证实该类化合物能够抑制微管蛋白的聚合,具有开发成新型抗肿瘤药物的潜能。
技术实现要素:
6.本发明的目的是提供一类含有2-芳基-4-取代喹啉结构的化合物,此类化合物具有全新的骨架结构和优异的抗肿瘤活性,能够用于制备抗肿瘤药物。
7.本发明的第二个目的是提供一种所述含有2-芳基-4-取代喹啉结构的化合物在制备抗肿瘤药物中的应用。
8.为了实现上述目的,本发明采用的技术方案如下:
9.本发明的第一方面提供了一类含有2-芳基-4-取代喹啉结构的化合物,结构如通式i所示:
[0010][0011]
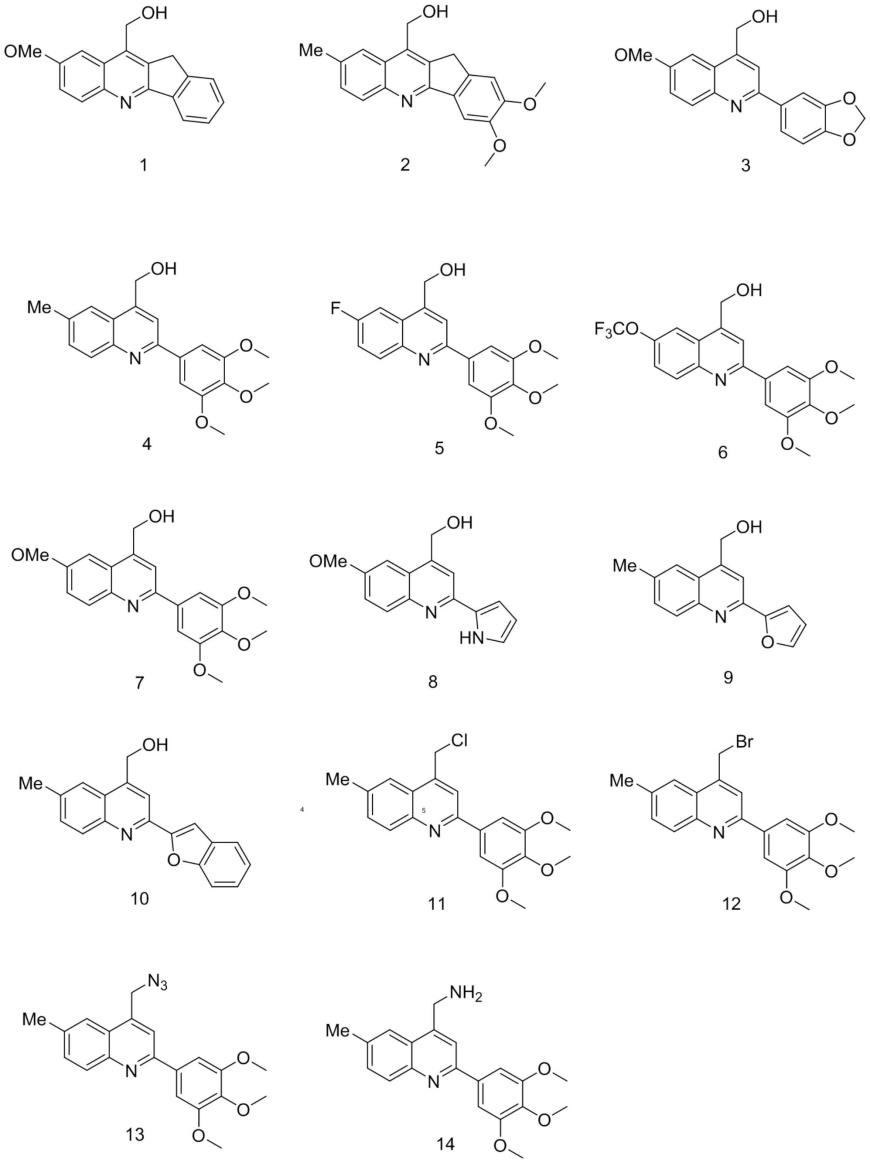
结构通式中y为-oh、-cl、-br、-n3、-nh2;r1和r2为-me、-ome、-f、-ocf3取代基团;喹啉的2位芳基可以为苯环或为杂环如吡咯、呋喃或苯并呋喃。
[0012]
结构式中的n为1代表一个碳原子相连,n为0代表该部分为断开结构。
[0013]
最优选的,所述通式i结构或其药用盐的结构选自以下结构的一种:
[0014][0015]
本发明的第二方面提供了一种所述2-芳基-4-取代喹啉结构的化合物或其药用盐在制备抗肿瘤药物中的应用。
[0016]
所述2-芳基-4-取代喹啉结构化合物优选为化合物1-14。其中最优选的为化合物4和6。
[0017]
所述肿瘤为肺癌、肠癌或乳腺癌。
[0018]
由于采用上述技术方案,本发明具有以下优点和有益效果:
[0019]
本发明提供的2-芳基-4-取代喹啉结构化合物,对乳腺癌mda-mb-231、肺癌a549和
肠癌hct116均产生了较为明显的增殖抑制作用,部分化合物的抗肿瘤活性强于临床药物顺铂。如化合物4总体表现最优的抗肿瘤活性,对肺癌、乳腺癌和肠癌的半数抑制浓度ic
50
分别为0.86μm,0.65μm和0.90μm,优于临床药物顺铂(ic
50
》5μm),可作为抗肿瘤的先导结构进行更加深入的研究。本发明提供的化合物具有全新的骨架结构,含有羟基、氨基等基团,有利于进一步地快速衍生化;较前期研究成果相比,这些化合物具有全新的骨架结构;而且这类化合物具有较强的抗肿瘤活性,可以进行抗肿瘤药物的开发。
[0020]
本发明提供的含有2-芳基-4-取代喹啉结构的化合物合成路线简单,合成原料易得、合成方法容易实现。
[0021]
本发明提供的含有2-芳基-4-取代喹啉结构的化合物在制备抗癌药物制剂中的应用,能够为临床治疗提供更多的选择。
[0022]
本发明提供的2-芳基-4-取代喹啉结构的化合物,具有更简便的化学合成路线,部分化合物较临床药物顺铂表现更优异的体外抗肿瘤活性。
附图说明
[0023]
图1a是化合物4对微管蛋白聚合抑制实验。
[0024]
图1b是秋水仙碱对微管蛋白聚合抑制实验。
[0025]
图1c是化合物4对微管蛋白秋水仙碱结合位点的分子对接图。
[0026]
图1d是化合物4与秋水仙碱在结合位点的叠合图。
具体实施方式
[0027]
为了更清楚地说明本发明,下面结合优选实施例对本发明做进一步的说明。本领域技术人员应当理解,下面所具体描述的内容是说明性的而非限制性的,不应以此限制本发明的保护范围。
[0028]
下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。
[0029]
以下实施例制备的化合物的化学结构式、1h-nmr、
13
c-nmr和hrms数据详见表1,其中编号1~14分别对应实施例1~14制备的化合物1~14。
[0030]
表1.目标化合物1~10的化学结构式、1h-nmr、
13
c-nmr和hrms数据
[0031]
[0032]
[0033]
[0034][0035]
实施例1
[0036]
化合物1的合成
[0037][0038]
步骤1:取5-甲氧基靛红(150mg,0.85mmol)和2,3-dihydro-1h-inden-1-one(112mg,0.85mmol)、koh(143mg,2.55mmol)加入15ml etoh在80℃下加热回流反应过夜。加入稀盐酸调ph到2。抽滤得到白色固体(189mg,收率57%).
[0039][0040]
取步骤1反应所得化合物(37mg,0.127mmol)和nabh4(14.2mg,0.375mmol)加入thf(3ml),将温度降至0℃搅拌并在1h内滴加bf3·
oet2(60μl),加热至室温反应16h。加入2ml 1%的氢氧化钠水溶液。搅拌3h。减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=50:1)进行纯化。得到化合物1,棕色固体(11mg,收率31%)
[0041]
实施例2
[0042]
化合物2的合成
[0043][0044]
参照实施例1,反应得到化合物2,棕色固体(12mg,收率为29%)。
[0045]
实施例3
[0046]
化合物3的合成:
[0047][0048]
步骤1:取5-甲氧基靛红(150mg,0.85mmol)和1-(benzo[d][1,3]dioxol-5-yl)ethan-1-one(140mg,0.85mmol)、koh(143mg,2.55mmol)加入15ml etoh在80℃下加热回流反应过夜。
[0049]
加入稀盐酸调ph到2。抽滤得到黄色固体(186mg,yield=68%)。
[0050][0051]
步骤2:取step1反应所得化合物(40mg,0.12mmol)在thf(3ml)中,在0℃搅拌并加入lialh4(0.24mmol),加热至室温反应30min.加入无水甲醇淬灭,减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=30:1)进行纯化。得到化合物3,黄色固体(27mg,收率73%)
[0052]
实施例4
[0053]
化合物4的合成
[0054][0055]
参照实施例3,反应得到化合物4,淡白色固体(27mg,收率为66%)。
[0056]
实施例5
[0057]
化合物5的合成
[0058][0059]
参照实施例3,反应得到化合物5,白色固体(21mg,收率为51%)。
[0060]
实施例6
[0061]
化合物6的合成
[0062][0063]
参照实施例3,反应得到化合物6,白色固体(20mg,收率为40%)。
[0064]
实施例7
[0065]
化合物7的合成
[0066][0067]
参照实施例3,反应得到化合物7,白色固体(25mg,收率为59%)。
[0068]
实施例8
[0069]
化合物8的合成
[0070][0071]
参照实施例3,反应得到化合物8,淡白色固体(25mg,收率为81%)。
[0072]
实施例9
[0073]
化合物9的合成
[0074]
[0075]
参照实施例3,反应得到化合物9,淡白色固体(24mg,收率为83%)。
[0076]
实施例10
[0077]
化合物10的合成
[0078][0079]
参照实施例3,反应得到化合物10,淡白色固体(18mg,收率为51%)。
[0080]
实施例11
[0081]
化合物11的合成
[0082][0083]
取化合物4(20mg,0.059mmol)加入5ml dcm,在0℃搅拌并加入socl2(172μl),反应3h。减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=25:1)进行纯化。得到化合物11,黄色固体(11mg,收率52%)
[0084]
实施例12
[0085]
化合物12的合成
[0086][0087]
取化合物4(40mg,0.12mmol)和nbs(43mg,0.24mmol)加入thf(3ml),在0℃搅拌并加入pph3(62mg,0.24mmol),加热到室温,反应12h。减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=25:1)进行纯化。得到化合物12,黄色固体(26mg,收率54%)。
[0088]
实施例13
[0089]
化合物13的合成
[0090][0091]
化合物12(20mg,0.05mmol)和nan3(6.5mg,0.1mmol)加入1ml dmf中,室温搅拌反应12h。减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=30:1)进行纯化。得到化合物13,棕色固体(11mg,收率60%)。
[0092]
实施例14
[0093]
化合物14的合成
[0094][0095]
化合物13(30mg,0.08mmol),pph3(44mg,0.17mmol)和h2o(200μl)加入3ml thf中,室温搅拌反应12h。减压蒸干溶剂,残留物用硅胶柱层析(dcm:meoh=30:1)进行纯化。得到化合物14,棕色固体(12mg,收率44%)。
[0096]
实施例15
[0097]
本发明化合物的抗肿瘤活性试验。
[0098]
对本发明的化合物进行肿瘤细胞增殖抑制试验,试验方法采用常规的ckk-8法。
[0099]
细胞株选用肺癌a549、结肠癌hct116、乳腺癌mda-mb-231,均购自上海生命科学研究院细胞库。培养液为dmem+10%nbs+双抗。
[0100]
样品液配制:待测化合物用dmso(merck)溶解后,配成浓度为10mm的母液。用培养基稀释母液,配成药物的最终浓度分别为100μm、50μm、25μm、12.5μm、6.25μm和3.125μm,1.56μm,0.78μm,0.39μm。
[0101]
96孔板每孔加入浓度为8
×
104个/ml的细胞悬液100μl,即8000个细胞/孔,置37℃、5%co2培养箱内。24小时后,吸掉上层培养液,加入含有样品的培养液和对照品液,100μl/孔,37℃作用72小时。每孔加入ckk-810μl,置培养箱内,作用1小时后用mk-2全自动酶标仪测570nm od值,计算半数抑制浓度ic
50
。
[0102]
部分优选化合物的抗肿瘤活性详见表2,其中,样品1~14是指相应实施例中制备的化合物,如化合物1表示在实施例1中所得到的化合物,同理类推。阳性药为秋水仙碱、顺铂。
[0103]
表2.本发明部分化合物对肿瘤细胞的半数抑制浓度ic
50
(单位:μm)
[0104][0105][0106]
表2中结果显示,本技术的多数化合物表现出较好的抗肿瘤活性,对肺癌a549、结肠癌hct116和乳腺癌mda-mb-231均产生了优异的增殖抑制作用。部分化合物的抗肿瘤活性优于顺铂,如化合物4、6、7对肺癌a549、结肠癌hct116和乳腺癌mda-mb-231的半数抑制浓度ic
50
都低于5μm,体现出了广谱,优异的抗肿瘤活性,尤其是化合物4,其对上述三种肿瘤细胞的半数抑制浓度ic
50
均低于1μm。此外,化合物3和化合物10对上述三种肿瘤细胞的半数抑制浓度ic
50
均低于12μm,产生了较为优异的抗肿瘤活性。结合化学结构分析,上述5种化合物具有以下共同点:1.y为-oh;2.n为0;3.喹啉的2位芳基含有苯环(苯环3,4,5位有三个甲氧基效果最好)。因此,具有以上所述结构特点的化合物骨架为本发明提供的含有2-芳基-4-取代喹啉结构的化合物产生优异抗肿瘤活性的优势结构骨架,可以进行抗肿瘤药物的开发。
[0107]
实施例16
[0108]
微管蛋白聚合抑制试验
[0109]
该实验参照我们已经发表的论文(bioorg chem,2022,118,105486)。将从猪的脑中分离的微管蛋白溶解在含有1mm gtp、5%甘油和化合物的pem缓冲液中。以秋水仙碱为阳性对照。用spectramax 190分光光度计在37℃下检测混合物在340nm处的吸光度。用稳定吸光度值进行计算。该试验进行了两次,并据此计算ic
50
值。
[0110]
如附图一所示:化合物4能够抑制微管蛋白的聚合(图1a),ic
50
为13.5μm。阳性药秋水仙碱的ic
50
为8.1μm(图1b)。分子对接结果显示,化合物4能够很好的插入微管蛋白的秋水仙碱结合位点,其羟基与氨基酸asn 101形成氢键相互作用,增强了结合(图1c)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1