一种构建牡蛎单倍型基因组的方法
:本发明属于海洋生物基因组和分子遗传学领域,具体涉及到一种构建牡蛎单倍型基因组的方法。
背景技术
0、
背景技术:
1、牡蛎等海洋贝类的基因组比较复杂,常规基因组组装策略无法获得高质量的单倍型基因组。随着测序技术的发展,测序的成本下降较快,数据的产出量和测序序列的长度均有所增加,但是一般情况下牡蛎等二倍体生物基因组的常规拼接组装无法获得两套全单倍型基因组序列,获得的是两套全单倍型基因组序列的马赛克嵌合形式,无法实现全基因组序列的定相,制约了基因组序列在单倍型鉴定、结构变异检测、比较基因组和连锁不平衡等遗传学分析中的应用。目前对牡蛎等贝类复杂基因组进行单倍型组装尚未有成熟有效的方法和策略。
技术实现思路
0、
技术实现要素:
1、本发明目的在于提供一种构建牡蛎单倍型基因组的方法。
2、为实现上述目的,本发明采用技术方案为:
3、一种构建牡蛎单倍型基因组的方法,
4、(1)选取牡蛎雌性和雄性个体,人工构建全同胞家系并对后代进行常规养殖;
5、(2)后代个体养殖一年后,取一个体进行长序列高通量测序;
6、(3)对全同胞家系的母本和父本个体分别进行短序列高通量测序;
7、(4)利用后代测序序列进行初步组装;
8、(5)将亲本序列比对到初步组装,鉴定亲本特异性单核苷酸变异位点;
9、(6)利用亲本特异性单核苷酸变异位点对后代个体的测序序列进行分组,获得父本来源和母本来源的长序列;
10、(7)将两个亲本来源的长序列分别进行组装,获得两个单倍型基因组序列。
11、所述步骤(1)在牡蛎繁殖季节期收集壳高>5cm的一龄或者二龄牡蛎雌性和雄性个体若干,待用;其中,雌、雄牡蛎可以是同种牡蛎,也可为不同种牡蛎。
12、所述步骤(2)取一个体进行长序列高通量测序;其中,长序列高通量测序采用pacbio公司的smart测序模式获得ccs序列,测序深度不低于60×。
13、所述步骤(3)中短序列高通量测序深度均不低于50×,而后对雌、雄亲本个体的基因组大小、杂合度和重复序列比例进行估算。所述基因组大小估算值(ge)是为后面序列组装获得的基因组大小(go)提供参考,ge大小无范围要求。杂合度估算值(hete)最好>1%,较低的杂合度不利于后面的基于snp的测序序列分组,重复序列比例值(rep)一般要求<70%,过高的rep值会导致基因组序列组装的碎片化。
14、所述步骤(4)以步骤2中获得的子代个体序列为输入,进行拼接,拼接后完整性需要达到complete buscos(c)指标值不低于85%。
15、将步骤(3)亲本的测序序列分别与步骤(4)获得基因序列进行比对,根据比对信息鉴定亲本特异性单核苷酸变异位点。
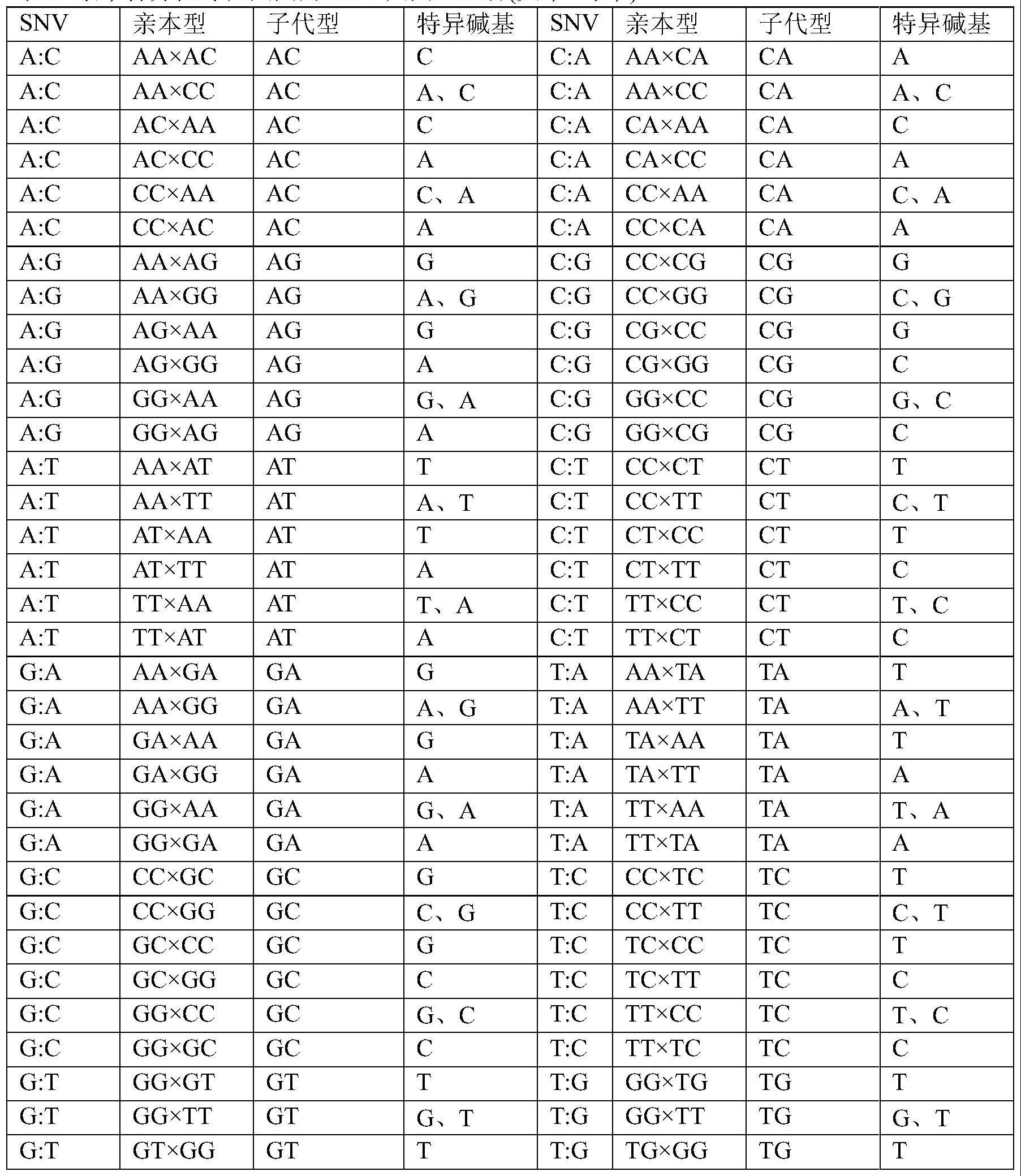
16、所述亲本特异性单核苷酸变异位点为在两个亲本中基因型不同,且后代中存在亲本特异性等位基因的snv基因型组合;其中,snv基因型组合形式:
17、(1)ff×mm,f为父本特异,m为母本特异;
18、(2)ff×fm,f不能判定来源,m为母本特异;
19、(3)fm×mm,f为父本特异,m不能判定来源;
20、其中f和m代表基因组dna四种组成碱基a、c、g和t。
21、具体实施过程如下:
22、1.针对待测牡蛎种类,在牡蛎繁殖季节期收集壳高>5cm的一龄或者二龄牡蛎个体若干。用牡蛎刀将牡蛎的双壳完全撬开后,挑选性腺发育饱满的个体,利用普通光学显微镜镜检性腺组织细胞,确定牡蛎性别。采用一雄对一雌人工授精的方法构建牡蛎全同胞家系,进行常规人工养殖。同时,将用于家系构建的亲本牡蛎个体的外套膜组织-80℃冻存备用。
23、2.全同胞家系子代个体养殖大约一年后,随机挑选壳高>5cm的个体一只,开壳后解剖,取外套膜组织约300-500mg,酚氯仿法抽提大片段基因组dna,质检合格后,利用pacbio smart sequelii测序仪进行测序。将下机原始数据的bam文件利用ccs程序进行处理,获得ccs序列。累加所有ccs序列的长度,计算子代序列的基因组覆盖度。如果覆盖度小于60×,则需要进行数据加测。
24、3.从雌雄亲本的外套膜组织中提取基因组dna序列,质检合格后,利用illuminahiseq系列或者novaseq6000等测序仪进行测序。将下机原始数据的fastq文件利用fastp程序进行过滤,去除平均质量值低于20的序列,累加高质量序列长度,计算序列的基因组覆盖度,要求达到50×以上,否则需要进行数据加测。利用genomescope程序对雌雄亲本和子代个体的基因组大小、杂合度和重复序列比例进行估算。
25、4.利用hifiasm等程序,以步骤2中获得的子代个体ccs序列为输入,进行拼接。初步拼接结束后,利用purgehaplotigs程序对组装序列中的冗余序列进行清除。利用busco程序以metazoa_odb10数据库为参照对组装的完整性进行评估,完整性需要达到completebuscos(c)指标值不低于85%。如果c值低于85%,表明步骤2中的测序数据量过低,需要加测数据。如果complete and duplicated buscos(d)指标值大于10%,表明存在过多的冗余序列,需要利用purgehaplotigs程序对组装序列进行多次去冗余。该步骤结束后,获得二倍体融合基因组组装序列集。
26、5.将亲本的测序序列分别利用bwa程序比对到步骤4获得的基因组序列,获得比对bam文件,利用samtools程序对bam文件中的序列进行排序,利用picard程序去除bam文件中的pcr扩增导致的重复比对,利用gatk程序对父本bam和母本bam文件进行单核苷酸变异(snv)检测,鉴定亲本特异性snv。具体标准为:在两个亲本中基因型不同,且后代中存在亲本特异性等位基因的snv基因型组合,包括3种模式:ff×mm、ff×fm和fm×mm。具体的基因型组合如下表1所示。
27、表1亲本特异性等位基因的snv基因型组合(父本×母本)
28、
29、
30、6.将子代个体ccs序列利用minimap2程序比对到步骤4获得的基因组序列,获得比对bam文件,将步骤5中获得的亲本特异性snv在子代ccs序列中进行定位,统计在子代ccs序列中该snv的等位基因碱基类型。根据表1中snv的亲本基因型和子代基因型的碱基组合形式,统计每一条子代ccs序列中测序质量值大于q20的父本特异碱基的数目和母本特异碱基的数目,确定序列来源。最终获得父本特异序列集和母本特异序列集。
31、7.利用hifiasm等程序,以步骤6中获得的两个亲本来源的子代ccs序列为输入,进行拼接。拼接结束后,如果基因组序列长度明显大于预期值,需要利用purgehaplotigs程序对组装序列中的冗余序列进行清除。利用busco程序以metazoa_odb10数据库为参照对组装的完整性进行评估,完整性需要达到complete buscos(c)指标值不低于90%。如果c值低于90%,表明步骤2中的测序数据量过低,需要加测数据。如果complete and duplicatedbuscos(d)指标值大于5%,表明存在过多的冗余序列,需要利用purgehaplotigs程序对组装序列进行多次去冗余。该步骤结束后,获得两个单倍型基因组。
32、本发明所具有的优点:
33、本发明单倍型基因组构建方法能够实现测序个体父源序列和母源序列的分组和拼接组装,从而同时获得父源和母源高质量单倍型基因组。应用该方法获得长牡蛎和福建牡蛎高质量单倍型基因组,busco评估基因组的完整性大于93%,表明该方法获得的牡蛎单倍型基因组质量较高。与普遍采用的混合组装策略相比,在测序深度较低的情况下,可以获得较高的序列连续性和完整性。
34、本发明采用鉴定亲本特异性snp并用于子代个体测序序列的分组和单独组装的策略,可以实现亲本单倍型测序序列的高效分组,充分发挥牡蛎高杂合度的优势,获得两个完全定相的单倍型基因组,分别为父本个体的单倍型基因组和母本个体的单倍型基因组。
- 还没有人留言评论。精彩留言会获得点赞!