一种3,3-双噻吩的合成方法与流程
1.本发明涉及噻吩类衍生物合成技术领域,尤其涉及一种3,3-双噻吩的合成方法。
背景技术:
2.3,3-双噻吩具有较高的空穴迁移率和电导率,良好的热稳定性和易于成膜的特点,是光电领域中最有潜力的光电材料之一,同时作为优良的材料中间体,在光催化降解有机污染物领域、生物电致变色材料领域、阳离子表面活性剂半导体封装领域和电磁屏蔽材料领域等存在广泛的应用价值。
3.传统的3,3-双噻吩合成方法有金属催化偶联法和化学氧化聚合法,金属催化偶联法以单卤代噻吩为原料在过渡金属钯催化下完成,但该方法产率低,实际应用价值不高;化学氧化聚合法通常是将单体溶于水溶液或有机溶液中,在酸性条件下加入合适的氧化剂反应,得到目标产物,但是这种方法工艺条件难以控制,氧化剂的种类与浓度、反应时间及温度、单体浓度以及酸性等工艺参数的微小变化都会对反应产物造成显著影响,导致该方法操作复杂。
技术实现要素:
4.针对以上问题,本发明的目的在于提供一种3,3-双噻吩的合成方法,本发明提供的合成方法操作简单,适合放大生产,得到的3,3-双噻吩产率高,纯度高。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种3,3-双噻吩的合成方法,包括以下步骤:
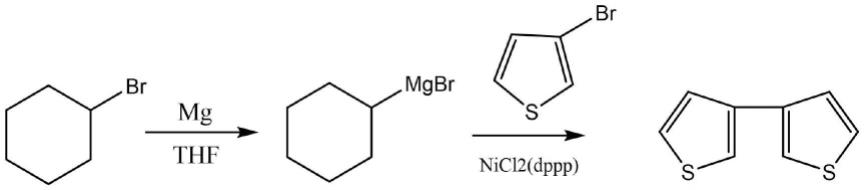
7.将环己基卤化镁、卤代噻吩、过渡金属催化剂和环烷烃类溶剂混合进行加成反应,得到3,3-双噻吩。
8.优选的,所述环己基卤化镁的制备方法为:将金属镁、醚类溶剂、催化剂和卤代环己烷混合进行格氏反应,得到环己基卤化镁。
9.优选的,所述金属镁为镁屑、镁条和镁粉中的一种或几种;所述卤代环己烷包括溴代环己烷、氯代环己烷和碘代环己烷中的一种;所述催化剂为碘、甲基碘化镁和乙基溴化镁中的一种或几种;所述醚类溶剂为四氢呋喃、乙醚和甲基叔丁基醚中的一种或几种。
10.优选的,所述过渡金属催化剂包括nicl2、ni(co)4、sncl2·
h2ptcl6和hco(co)4中的一种或几种;所述卤代噻吩包括3-溴噻吩、3-氯噻吩和3-碘噻吩中的一种。
11.优选的,所述环烷烃类溶剂为环己烷和环戊烷中的一种或两种。
12.优选的,所述卤代环己烷与金属镁的摩尔比为1:1.5~1;所述卤代环己烷与催化剂的摩尔比为1:0.005~0.03。
13.优选的,所述环己基卤化镁与卤代噻吩的摩尔比为1.5~1:1,所述卤代噻吩与过渡金属催化剂的摩尔比1:0.005~0.05。
14.优选的,所述格氏反应的温度为-78~70℃,时间为4~6h。
15.优选的,所述加成反应的温度为-78~70℃,时间为40~90min。
16.优选的,所述加成反应终止后还包括:对所得反应液进行提纯;所述提纯为将反应液与提纯用溶剂搅拌混合后静置分层,得到含3,3-双噻吩的有机层,对含3,3-双噻吩的有机层依次进行水洗、干燥、脱溶浓缩和重结晶,得到3,3-双噻吩。
17.本发明提供了一种3,3-双噻吩的合成方法,包括以下步骤:将环己基卤化镁、卤代噻吩、过渡金属催化剂和环烷烃类溶剂混合进行加成反应,得到3,3-双噻吩。本发明利用环己基卤化镁与卤代噻吩完成加成反应,生成3,3-双噻吩。本发明提供的合成方法操作简便,且工艺简单,基本无副反应产生。
18.本发明提供的合成方法工艺简便、反应高效,同时该合成方法的目标产物纯度高,基本无危废产生,适合放大生产,因此能够促进3,3-双噻吩的商业化进程,批量生产的3,3-双噻吩可作为重要药物中间体,可用于有机光电材料的研发与生产,有力带动国内外双噻吩类衍生物材料的使用规模,具有显著的社会效益。
具体实施方式
19.本发明提供了一种3,3-双噻吩的合成方法,包括以下步骤:
20.将环己基卤化镁、卤代噻吩、过渡金属催化剂和环烷烃类溶剂混合进行加成反应,得到3,3-双噻吩。
21.在本发明中,若无特殊说明,所述各物质均为本领域技术人员熟知的市售商品。
22.在本发明中,所述环己基卤化镁(rmgx,r为环己基)的制备方法优选为:将金属镁、醚类溶剂、催化剂和卤代环己烷混合进行格氏反应,得到环己基卤化镁。在本发明中,所述金属镁优选为镁屑、镁条和镁粉中的一种或几种,更优选为镁屑;所述卤代环己烷优选包括溴代环己烷、氯代环己烷和碘代环己烷中的一种,更优选为溴代环己烷;所述催化剂优选为碘、甲基碘化镁和乙基溴化镁中的一种或几种,更优选为碘;所述醚类溶剂优选为四氢呋喃、乙醚和甲基叔丁基醚中的一种或几种,更优选为无水四氢呋喃、无水乙醚和无水甲基叔丁基醚中的一种或几种;所述卤代环己烷与金属镁的摩尔比优选为1:1.5~1;所述卤代环己烷与催化剂的摩尔比优选为1:0.005~0.03。
23.在本发明中,所述混合优选为:在常温常压下将金属镁、醚类溶剂和催化剂进行第一混合,得到第一混合物,在搅拌条件下,将所述第一混合物缓慢升温至30~70℃,将所述第一混合物与部分卤代环己烷进行第二混合,得到第二混合物,待反应引发后,将剩余卤代环己烷滴加至所述第二混合物中;所述卤代环己烷优选以卤代环己烷与醚类溶剂的混合溶液形式进行混合;所述醚类溶剂优选为四氢呋喃、乙醚和甲基叔丁基醚中的一种或几种,更优选为四氢呋喃;所述格氏反应的温度优选为-78~70℃,更优选为-50~50℃,进一步优选为30~40℃,时间优选为4~6h,所述格氏反应的时间自剩余卤代环己烷开始滴加时计算。在本发明的具体实施例中,所得含有环己基卤化镁的反应液无需处理,直接进行下一步加成反应。
24.在本发明中,所述过渡金属催化剂优选包括nicl2、ni(co)4、sncl2·
h2ptcl6和hco(co)4中的一种或几种,更优选为nicl2;所述卤代噻吩包括3-溴噻吩、3-氯噻吩和3-碘噻吩中的一种,更优选为3-溴噻吩;所述环烷烃类溶剂优选为环己烷和环戊烷中的一种或两种;所述环己基卤化镁与卤代噻吩的摩尔比优选为1.5~1:1,所述卤代噻吩与过渡金属催化剂的摩尔比优选为1:0.005~0.05;所述混合优选为:先将所述环己基卤化镁和过渡金属催化
剂进行第三混和,得到第三混合液,再将卤代噻吩和环烷烃类溶剂的混合溶液滴加至第三混合液中;所述加成反应的温度优选为-78~70℃,更优选为-50~50℃,进一步优选为30~40℃,时间优选为40~90min,更优选为50~80min,进一步优选为50~60min;所述加成反应的时间自卤代噻吩和环烷烃类溶剂的混合溶液开始滴加时计算。
25.在本发明中,达到反应时间后,本发明优选终止反应,然后对所得反应液进行提纯;所述终止反应用溶液优选为氯化铵饱和水溶液;所述氯化铵饱和水溶液与反应液的体积之比优选为0.5~1.5:1;所述提纯优选为将反应液与提纯用溶剂搅拌混合后静置分层,得到含3,3-双噻吩的有机层,对含3,3-双噻吩的有机层依次进行水洗、干燥、脱溶浓缩和重结晶,得到3,3-双噻吩;所述提纯用溶剂优选为乙酸乙酯、乙酸甲酯和二氯甲烷中的一种或几种;所述水洗的用水量优选为500~1000ml;所述水洗次数优选为2次;所述干燥用试剂优选为无水硫酸钠;所述脱溶浓缩优选为将干燥后的含3,3-双噻吩的有机层进行旋蒸直至无溶剂蒸出;所述旋蒸的温度优选为40℃;本发明对所述重结晶无特殊要求,选择本领域技术人员常用的方式即可。
26.本发明合成3,3-双噻吩的反应路线如下:
[0027][0028]
本发明首先利用格氏反应高效、温和的特点,在反应中引入格氏试剂生成环己基卤化镁,再与卤代噻吩完成加成反应,生成3,3-双噻吩。
[0029]
本发明提供的3,3-双噻吩的合成方法工艺简单,合成得到的3,3-双噻吩纯度高达98%以上,收率高达61%以上。
[0030]
为了进一步说明本发明,下面结合实施例对本发明提供的一种3,3-双噻吩的合成方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0031]
实施例1
[0032]
在常温常压下,依次将30.7g镁屑、50ml无水四氢呋喃与0.13g碘放入反应容器中混合均匀,在搅拌条件下,缓慢升温至70℃,滴加10ml溴代环己烷和无水四氢呋喃的混合溶液,其中所述混合溶液由167g溴代环己烷与500ml无水四氢呋喃组成,待反应开始引发后,保持体系温度70℃,继续滴加剩余的溴代环己烷和无水四氢呋喃的混合溶液,反应4h后,保持体系温度为70℃,加入2gnicl2(dppp),滴加163.3g 3-溴噻吩与300ml环己烷的混合溶液,反应70min,得到反应液,向所得反应液中依次加入1l氯化铵饱和水溶液和1l乙酸乙酯,继续搅拌30s,静置分层,将所得有机层水洗两次,每次用水500ml,然后将有机层用无水硫酸钠干燥,过滤除去无水硫酸钠,将除水后的有机层进行旋蒸直至无乙酸乙酯,旋蒸的温度为40℃,所得剩余物进行重结晶,得到50.8g白色片状晶体3,3-双噻吩,纯度为98%,收率为61%。
[0033]
实施例2
[0034]
在常温常压下,依次将31.2g镁屑、50ml无水乙醚与0.1g碘放入反应容器中混合均匀,在搅拌条件下,缓慢升温至40℃,滴加10ml溴代环己烷和无水乙醚的混合溶液,其中所
述混合溶液由163g溴代环己烷与500ml无水乙醚组成,待反应开始引发后,保持体系温度40℃,继续滴加剩余的溴代环己烷和无水乙醚的混合溶液,反应4h后,保持体系温度为40℃,加入2gnicl2(dppp),滴加163g 3-溴噻吩与300ml环戊烷的混合溶液,反应60min,得到反应液,向所得反应液中依次加入1l氯化铵饱和水溶液和1l乙酸乙酯,继续搅拌30s,静置分层,将所得有机层水洗两次,每次用水500ml,然后将有机层用无水硫酸钠干燥,过滤除去无水硫酸钠,将除水后的有机层进行旋蒸直至无乙酸乙酯,旋蒸的温度为40℃,所得剩余物进行重结晶,得到52.5g白色片状晶体3,3-双噻吩,纯度为98%,收率为63%。
[0035]
实施例3
[0036]
在常温常压下,依次将31.2g镁屑、50ml无水四氢呋喃与0.15g碘放入反应容器中混合均匀,在搅拌条件下,缓慢升温至35℃,滴加10ml溴代环己烷和无水四氢呋喃的混合溶液,其中所述混合溶液由164.5g溴代环己烷与500ml无水四氢呋喃组成,待反应开始引发后,保持体系温度35℃,继续滴加剩余的溴代环己烷和无水四氢呋喃的混合溶液,反应4h后,保持体系温度为35℃,加入2gnicl2(dppp),滴加163g 3-溴噻吩与300ml环戊烷的混合溶液,反应60min,得到反应液,向所得反应液中依次加入1l氯化铵饱和水溶液和1l乙酸乙酯,继续搅拌30s,静置分层,将所得有机层水洗两次,每次用水500ml,然后将有机层用无水硫酸钠干燥,过滤除去无水硫酸钠,将除水后的有机层进行旋蒸直至无乙酸乙酯,旋蒸的温度为40℃,所得剩余物进行重结晶,得到51.5g白色片状晶体3,3-双噻吩,纯度为98.2%,收率为62%。
[0037]
实施例4
[0038]
在常温常压下,依次将30.2g镁屑、50ml无水甲基叔丁基醚与0.09g碘放入反应容器中混合均匀,在搅拌条件下,缓慢升温至35℃,滴加10ml溴代环己烷和无水甲基叔丁基醚的混合溶液,其中所述混合溶液由163g溴代环己烷与500ml无水甲基叔丁基醚组成,待反应开始引发后,保持体系温度35℃,继续滴加剩余的溴代环己烷和无水甲基叔丁基醚的混合溶液,反应4h后,保持体系温度为35℃,加入2gnicl2(dppp),滴加163g 3-溴噻吩与300ml环己烷的混合溶液,反应40min,得到反应液,向所得反应液中依次加入1l氯化铵饱和水溶液和1l乙酸乙酯,继续搅拌30s,静置分层,将所得有机层水洗两次,每次用水500ml,然后将有机层用无水硫酸钠干燥,过滤除去无水硫酸钠,将除水后的有机层进行旋蒸直至无乙酸乙酯,旋蒸的温度为40℃,所得剩余物进行重结晶,得到52.4g白色片状晶体3,3-双噻吩,纯度为98%,收率为63%。
[0039]
以上所述仅是本发明的优选实施方式,并非对本发明作任何形式上的限制。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1