一种砜吡草唑的合成方法与流程
1.本发明涉及一种砜吡草唑的合成方法,具体涉及一种合成过程简单、操作简单的砜吡草唑的合成方法,属于有机合成技术领域。
背景技术:
2.砜吡草唑是日本组合化学株式会社开发的,化学名为:3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑,具有如下所示的结构:砜吡草唑可用于大多数作物田的芽前土壤处理剂,施用后被杂草幼根与幼芽吸收,抑制幼苗早期生长,破坏分生组织与胚芽鞘,是植物体内vlcfa(极长侧链脂肪酸)(c20~c30)生物合成中严重的潜在抑制剂。
[0003]
砜吡草唑3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑,有以下现有技术报道:专利cn102666503中公开了其可采用以下路径合成:具体包括以下步骤:在碱性条件下,1-甲基-3-(三氟甲基)-1h-吡唑-5-醇、甲醛水溶液与5,5-二甲基-4,5-二氢异恶唑硫脒盐酸盐在水中进行缩合反应,再与氟利昂烷基化,得到砜吡草唑中间体3-[[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基]甲基硫烷基]-5,5-二甲基-4h-1,2-噁唑,再与双氧水氧化得到砜吡草唑3-[5-(二氟甲氧基)-1-甲基-3-(三氟甲基)吡唑-4-基甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-异噁唑。但是,该合成方法中存在产物收率过低、存在含硫废水、三废难以处理等缺点。
[0004]
专利cn101213181a中报道了以下路线合成砜吡草唑关健中间体的方法:上述方法在碱性条件下发生取代反应,但由于底物活性和水体系溶解度差的原因,导致反应收率很低,低于70%。
技术实现要素:
[0005]
针对现有技术存在的不足,本发明提供了一种砜吡草唑的合成方法,该方法为砜吡草唑的合成提供了新的路线,该路线反应步骤少、反应速度快、合成过程简单、产品容易分离,且反应过程中产生的三废少,环保性好,产物的纯度和收率也较高,适合工业化应用。
[0006]
本发明具体技术方案如下:一种砜吡草唑的合成方法,砜吡草唑的结构式如下式c所示:上述合成方法中,砜吡草唑是由式a所示的化合物a和式b所示的化合物b(3-溴-5,5-二甲基-4,5-二氢异噁唑)在催化剂、配体和碱的作用下反应得到;反应式如下:进一步的,上述合成方法中,化合物a与化合物b在催化剂条件下发生偶联反应。化合物a和化合物b均可以从市场上购买得到,或者也可以根据现有技术中报道的方法自行合成。
[0007]
进一步的,上述合成方法中,所述催化剂为钯催化剂或者铜催化剂。其中,所述钯催化剂为pd2(dba)3或pd(dba)2,其中,pd2(dba)3为三(二亚苄基丙酮)二钯,pd(dba)2为双二亚苄基丙酮钯。钯催化剂中,钯为零价态。所述铜催化剂为cui或(cuotf)2·
phh。
[0008]
进一步的,上述合成方法中,钯催化剂与膦配体搭配使用,铜催化剂与胺类配体搭配使用。所述膦配体为联苯类单膦配体,所述联苯类单膦配体可以为johnphos(2-(二叔丁基膦)联苯)、xphos(2-二环己基磷-2',4',6'-三异丙基联苯)、davephos(2-二环己膦基-2'-(n,n-二甲胺)-联苯)、sphos(2-双环己基膦-2',6'-二甲氧基联苯)、ruphos(2-双环已
基膦-2 ',6'-二异丙氧基联苯)、brettphos(2-(二环己基膦)3,6-二甲氧基-2
′
,4
′
,6
′‑
三异丙基-1,1
′‑
联苯)等。所述胺类配体可以为n,n
′‑
二甲基乙二胺、l-proline(l-脯氨酸)等。
[0009]
进一步的,上述合成方法中,采用钯催化剂时,化合物a与钯催化剂的摩尔比为1:0.5%~1%,例如1:0.5%、1:0.6%、1:0.7%、1:0.8%、1:0.9%、1:1.0%。化合物a与膦配体的摩尔比为1:1.5%~4%,例如1:1.5%、1:1.8%、1:2%、1:2.2%、1:2.4%、1:2.6%、1:2.8%、1:3%、1:3.2%、1:3.4%、1:3.6%、1:3.8%、1:4%。采用铜催化剂时,化合物a与铜催化剂的摩尔比为1:0.1%~0.5%,例如1:0.1%、1:0.2%、1:0.3%、1:0.4%、1:0.5%。化合物a与胺类配体的摩尔比为1:1%~1.5%,例如1:1%、1:1.1%、1:1.2%、1:1.3%、1:1.4%、1:1.5%。
[0010]
进一步的,上述合成方法中,当催化剂为钯催化剂时,所用的碱为碳酸钠、碳酸钾、磷酸钾或碳酸铯,优选为碳酸铯。采用铜催化剂时,所用的碱为碳酸钾、碳酸铯、磷酸钾、氢氧化钠、氢氧化钾、dbu(1,8-二氮杂二环十一碳-7-烯)、dabco(三乙烯二胺),优选为dabco或dbu。
[0011]
进一步的,当碱为无机碱时,使用时先将其研磨。
[0012]
进一步的,上述合成方法中,化合物a与碱的摩尔比为1:1.05~1:1.3,例如1:1.05、1:1.1、1:1.2、1:1.3。
[0013]
进一步的,上述合成方法中,化合物a与化合物b的摩尔比为理论摩尔比1:1。
[0014]
进一步的,上述合成方法中,反应在溶剂中进行,催化剂为钯催化剂时,所述溶剂为甲醇、乙醇、乙腈、丙酮、dmf、dmso或醚类溶剂,所述醚类溶剂为二氧六环、四氢呋喃(thf)、乙二醇二甲醚(dme)等。dmf为溶剂时,安全性相对较高,且产品收率和纯度也更高,因此优选为dmf。
[0015]
进一步的,上述合成方法中,催化剂为铜催化剂时,所述溶剂为甲醇、乙醇、乙腈、丙酮、dmf或dmso,优选为dmf或dmso。
[0016]
进一步的,上述合成方法中,催化剂为钯催化剂时,反应温度为80℃~110℃,例如80℃、90℃、100℃、110℃,优选为80-90℃。在此反应温度下,化合物a和化合物b在催化剂、配体和碱的存在下能迅速发生反应,反应时间<4小时。
[0017]
进一步的,上述合成方法中,催化剂为铜催化剂时,反应温度为100℃~130℃,例如100℃、110℃、120℃、130℃,优选为100-110℃。在此反应温度下,化合物a和化合物b在催化剂、配体和碱的存在下能发生反应,反应时间<24小时。
[0018]
进一步的,上述合成方法中,具体的反应步骤为:反应时,将化合物a与溶剂混合,然后加入催化剂、配体、碱、化合物b,升温进行反应。反应结束后,过滤反应液,所得滤液用水洗涤,然后分液取有机相,将有机相回收溶剂,即得产物。本发明后处理简单,易于操作,所得产品纯度高,收率高。
[0019]
进一步的,上述合成方法中,整个反应在气体保护下进行,所述保护性气体可以是氮气,也可以是氩气等惰性气体。
[0020]
本发明提供了一种合成砜吡草唑的新方法,利用化合物a与3-溴-5,5-二甲基-4,5-二氢异噁唑直接一步反应得到砜吡草唑。相对于现有的合成方法,本发明具有以下优点:(1)本发明反应条件温和,无需高压,对设备要求不高;(2)本发明反应步骤少、反应速度快、合成过程简单,通过工艺条件的选择,反应选
择性高、反应收率高,产品纯度高,经验证,在优选的工艺条件下,产品收率和纯度最高可达90%以上;(3)本发明得到的产品容易分离,后处理简单粗放,适合大规模生产;(4)本发明合成过程中产生的三废少、易处理,环保性好。
附图说明
[0021]
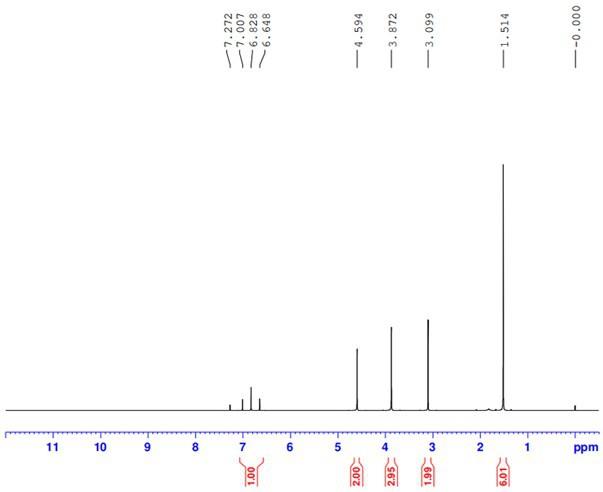
图1为实施例1中合成的砜吡草唑的核磁氢谱图。
具体实施方式
[0022]
下面结合具体的实施例对本发明的技术方案做进一步的描述,但本发明并不限于下述实施例。如果没有特殊说明,本发明采用的原料均可在市场上购买得到。或者也可以根据现有技术中报道的方法自行合成。
[0023]
实施例1室温条件下,向100mldmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.072g催化剂pd2(dba)3、0.11g配体xphos、5.67g碳酸铯、2.82g化合物b,升温至80-90℃反应,保温反应3.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物6.1g,经hplc检测,其纯度为91.0%,收率以化合物a计为89.70%。图1为产物的核磁氢谱图,其核磁信息如下:1h nmr(cdcl3):6.82(1h,t),4.59(2h,s),3.87(3h,s),3.09(2h,s),1.51(6h,s).实施例2室温条件下,向100ml甲醇中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.072g催化剂pd2(dba)3、0.11g配体xphos、1.84g碳酸钠、2.82g化合物b,升温至80-90℃反应,保温反应10.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物5.41g,经hplc检测,其纯度为89.8%,收率以化合物a计为78.5%。
[0024]
实施例3室温条件下,向100ml乙腈中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.072g催化剂pd2(dba)3、0.11g配体xphos、2.40g碳酸钾、2.82g化合物b,升温至80-90℃反应,保温反应6.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物5.70g,经hplc检测,其纯度为90.3%,收率以化合物a计为83.2%。
[0025]
实施例4室温条件下,向100mldmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.045g催化剂pd(dba)2、0.093g配体davephos、5.67g碳酸铯、2.82g化合物b,升温至80-90℃反应,保温反应3.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物6.2g,经hplc检测,其纯度为91.2%,收率以化合物a计为91.4%。
[0026]
实施例5室温条件下,向100ml dmso中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂
中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、1.95g dabco、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物5.6g,经hplc检测,其纯度为90.5%,收率以化合物a计为81.9%。
[0027]
实施例6室温条件下,向100ml 乙醇中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、1.95g dabco、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物3.2g,经hplc检测,其纯度为87.2%,收率以化合物a计为45.1%。
[0028]
实施例7室温条件下,向100ml 乙腈中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、1.95g dabco、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物4.8g,经hplc检测,其纯度为90.5%,收率以化合物a计为70.2%。
[0029]
实施例8室温条件下,向100ml dmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、1.95g dabco、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物5.9g,经hplc检测,其纯度为90.1%,收率以化合物a计为85.9%。
[0030]
实施例9室温条件下,向100ml dmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、2.64g dbu、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物6.0g,经hplc检测,其纯度为89.4%,收率以化合物a计为86.7%。
[0031]
实施例10按照实施例4的方法制备砜吡草唑,不同的是:所用的配体及其质量不同,具体见下表1所示。
[0032]
实施例12室温条件下,向100ml dmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入催化剂、配体、碱、2.82g化合物b,升温至100-110℃反应,保温反应20.0hr,hplc检测反应完全后,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物。
[0033]
催化剂、配体、碱的情况以及所得产品的收率和纯度如下表2所示。
[0034]
实施例13按照实施例4的方法制备砜吡草唑,不同的是:所用溶剂不同,具体见下表3所示。
[0035]
对比例1室温条件下,向100mldmf中加入5.0g化合物a搅拌;氮气保护的条件下,不加催化剂和配体,加入5.67g碳酸铯、2.82g化合物b,升温至80-90℃反应,保温反应24.0hr,hplc检测反应不完全,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物1.7g,经hplc检测,其纯度为67.2%,收率以化合物a计为18.5%。
[0036]
对比例2室温条件下,向100ml dmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.0086g催化剂cui、0.018g配体n,n
′‑
二甲基乙二胺、2.82g化合物b,不加碱,升温至100-110℃反应,保温反应20.0hr,hplc检测反应不完全,先将反应液热过滤,然后向滤液滴加入10g水,缓慢降温结晶,过滤后得到产物1.1g,经hplc检测,其纯度为56.1%,收率以化合物a计为9.97%。
[0037]
对比例3室温条件下,向100mldmf中加入5.0g化合物a搅拌;氮气保护的条件下,向溶剂中再加入0.072g催化剂pd2(dba)3、0.11g配体xphos、2.82g化合物b,不加碱,升温至80-90℃反应,保温反应24.0hr,hplc检测,几乎不反应。
[0038]
本发明已通过优选的实施方式进行了详尽的说明。然而,通过对前文的研读,对各实施方式的变化和增加也是本领域的一般技术人员所显而易见的。申请人的意图是所有这些变化和增加落在了本发明权利要求的保护范围中。本文中使用的术语仅为对具体的实施例加以说明,其并非意在对本发明进行限制。除非另有定义,本文中使用的所有术语(包括技术术语和科学术语)均与本发明所属领域的一般技术人员的理解相同。任何对此产品进行的修饰与改良,在专利范围或范畴内同类或相近物质的替代与使用,均属于本发明专利保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1