一种疏水缔合型阴离子聚丙烯酰胺及其制备方法和应用与流程
1.本发明属于高分子合成技术领域,具体涉及一种疏水缔合型阴离子聚丙烯酰胺的制备方法,且还涉及所述方法制备的疏水缔合型阴离子聚丙烯酰胺及其应用。
背景技术:
2.随着我国国民经济的飞速发展,印刷包装行业的快速增长带动了油墨制造业的快速成长,使得我国在最近的二十年间快速崛起成为世界油墨制造大国之一,特别是从近几年的生产状况来看,我国油墨生产水平整体保持稳中略升的进展态势,正以10%以上的速度持续快速增长。油墨行业分析指出,2022年中国油墨产量高达76.8万吨,其是我国轻工业的重要组成部分,是国民经济重要产业之一,在经济和社会进展中起着不容忽视的作用。然而,在油墨产业中,常规溶剂型油墨所使用的挥发性有机化合物会引起许多安全问题,如食品安全、环境污染、运输安全等,特别是在环境保护方面解决油墨制造和使用过程中对环境的污染问题成为目前最为迫切的课题,很多发达国家都制定了严格的法规,建立强有力的管制与监测机构,并不断地讨论开发各种各样的有效解决方法。
3.水性油墨的研究和应用范围正稳步增长,未来将逐渐取代溶剂型油墨的使用,我国水性油墨的研究已成为了油墨行业未来发展的重点方向。特别地,凹版印刷因其流动性好、内聚力强、干燥快速、能适应不规则承印物表面的印刷等优点逐渐成为水性油墨最重要的应用领域之一。现有的水性凹版印刷油墨为了提高其稳定性,通常会加入缔合型增稠剂,如cn109021686a公开了一种水性油墨及其制备方法,该水性油墨包括如下重量份数的组分:水性丙烯酸树脂30-50份;水性无树脂颜料15-20份;纯水3-5份;增稠剂2-5份;聚乙烯醇1-2份;γ-氨基丙基三乙氧基硅烷2-3份;表面活性剂2-6份;水性消泡剂0.01-0.015份;介孔材料0.3-0.6份;水性无树脂颜料的粒径为0.02-0.03μm;增稠剂包括羟丙基二淀粉磷酸酯、水性缔合型聚氨酯增稠剂中的至少一种;聚乙烯醇的聚合度为1200
±
200;表面活性剂包括非离子表面活性剂、阴离子表面活性剂中的至少一种;介孔材料包括介孔碳材料、介孔二氧化硅中的至少一种,该水性油墨采用疏水缔合型聚氨酯增稠剂,但这种增稠剂与油墨体系不能很好的兼容,特别是疏水缔合型聚氨酯难以调节其疏水基的微嵌段长度,增稠效果不佳,难以满足要求。
4.另外,为了提高水性油墨的印刷适应性,还会在其中添加醇类分散介质,如cn108250831a公开了一种环保水性凹版油墨及其制备方法,其利用醇水混合液将金属颜料、水性硅烷、碳酸钙和滑石粉润湿,反复碾磨后得到油墨色浆,添加流平剂、分散剂和消泡剂,低速搅拌制得油墨混合料,加入乳化树脂、水性蜡、硝酸锆、聚磺酸电解质,高速搅拌乳化后过滤,即得环保水性凹版油墨。cn11363403a公开了一种水性薄膜凹印复合油墨及其制备方法和应用,其水性薄膜凹印复合油墨以环保的水性聚氨酯树脂和水性丙烯酸树脂作为油墨的连接料,水性聚氨酯树脂和水性丙烯酸树脂在性能上具有互补作用,降低成本的同时克服彼此的缺陷。在凹版印刷水性油墨领域中,虽然采用水和醇作为分散介质满足了现有标准《gb/t26394-2011水性薄膜凹印复合油墨》中对于醇类溶剂的要求,但仍然会导致油
墨中有机溶剂含量仍然占有一定的比例(>10%),对大气环境存在不良影响。
5.综上,针对中国油墨制造业的快速发展期,如何改善水性油墨的稳定性并尽可能降低有机醇类物质的使用将会为油墨行业的进一步发展提供强有力的技术支撑。
技术实现要素:
6.为了解决现有技术中存在的问题,本发明对现有水性油墨增稠剂进行大量研究后提供一种新型疏水缔合型阴离子聚丙烯酰胺,其作为水性油墨的增稠剂使用时,兼具聚氨酯和聚丙烯酸两者的优势,具有良好的增稠性、抗剪切性和流动性,能够提高水性油墨在生产、储运和印刷过程中的稳定性。
7.在本发明的第一方面,本发明提供一种疏水缔合型阴离子聚丙烯酰胺,其结构式如式i所示:
[0008][0009][0010]
其中,变量a、b、c相同或不同且各自彼此独立,变量a是从43至22000的整数,变量b是从4至2000的整数,变量c是从2至960的整数,
[0011]
变量r1和r2相同或不同且各自独立地为氢、卤素、烷基、取代烷基、芳基、芳烷基、氰基。
[0012]
本发明疏水缔合型阴离子聚丙烯酰胺中引入亲水嵌段极性强且水溶性好,从而使其在用于水性油墨中能够为水作为分散介质提供很好的基础,且磺酸基的静电排斥力,能够使所获得聚丙烯酰胺不呈现卷曲状态,聚合物分子链更加伸展,疏水缔合作用加强。另外,本发明疏水缔合型阴离子聚丙烯酰胺中引入刚性疏水嵌段,其以微嵌段的形式分布于分子链上,使得所获得的聚合物在水溶液中易形成分子间缔合而具有良好的增粘性,且还能够有效提高聚丙烯酰胺的稳定性,使其在高温、高剪切下保持着较高的表观粘度,且对油墨体系没有副作用。
[0013]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,变量a、b、c的数值比例为(20-30):(2-5):1。所述变量a、b、c在合适的范围内才能保证本发明疏水缔合型阴离子聚丙烯酰胺在水溶液中通过静电相互作用、氢键相互作用或者范德华力作用或者这些作用力之间产生具有一定强度且又可逆的物理缔合,从而使水性油墨形成均一稳定的体系。
[0014]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,变量r1和r2中的一个代表h,另一个代表卤素、烷基、取代的烷基、芳基、芳烷基、氰基。
[0015]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,所述烷基为碳原子数为1至51
的烷基,更优选碳原子数为1-25的烷基,最优选碳原子数为1-10的烷基如甲基、乙基、丙基、丁基、戊基、己基等等。所述烷基可以为支链的或非支链的。
[0016]
在本发明中,所述烷基的碳原子数不可太多,原因在于碳原子太多,则疏水性太强,进而影响本发明疏水缔合型阴离子聚丙烯酰胺的水溶性。
[0017]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,所述取代烷基为被以下基团所取代的烷基:卤素、烷基、氰基、苄基、硝基。
[0018]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,所述芳基为具有单环的碳原子数为6-14个的芳香族碳环基团如苯基,进一步优选地,所述芳基可以为取代的或未取代的芳基,最优选的,所述芳基为苯基或者被卤素取代的苯基。
[0019]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,所述芳烷基是指式-烷基-芳基的基团,其中烷基和芳基是按上面定义的。所述芳烷基可以是取代的或未取代的,其中取代的芳烷基可以在该基团的烷基、芳基或者烷基和芳基部分处被取代。代表性的芳烷基包括但不限于苄基和苯乙基。
[0020]
优选地,在本发明疏水缔合型阴离子聚丙烯酰胺中,所述卤素为氟、氯、溴或者碘。卤素作为憎水基团,通过其取代可以进一步增加本发明疏水缔合型阴离子聚丙烯酰胺的疏水性,特别是f原子的取代,可以降低表面能,可以很好地提高本发明疏水缔合型阴离子聚丙烯酰胺的疏水性。
[0021]
优选地,本发明疏水缔合型阴离子聚丙烯酰胺的粘均分子量为5000-2000000,更优选500000-2000000,本发明疏水缔合型阴离子聚丙烯酰胺分子量可以通过调节制备过程中的各种参数来控制大小,进而实现不同场景下的使用选择,为使用者提供更优的选择。
[0022]
本发明粘均分子量采用gb12005.1-89“聚丙烯酰胺特性粘数测定方法”中规定的测试方法进行测定,采用氯化钠溶液做溶剂,并制备一定浓度的试样溶液,使用气承液柱式乌式粘度计,分别测定溶液和溶剂的流经时间,根据测定的值计算特性粘数,进而算出粘均分子量。
[0023]
本发明疏水缔合型阴离子聚丙烯酰胺可以通过调整单体种类和聚合物结构参数实现聚合物的分子量可控,且可达到百万级别,相比于增稠剂聚氨酯和聚丙烯酸更有优势,更能满足多样化需求。
[0024]
在本发明的第二方面,本发明提供一种制备上述疏水缔合型阴离子聚丙烯酰胺的方法,其包括以下步骤:
[0025]
(s1)将丙烯酰胺和2-丙烯酰胺-2-甲基丙磺酸溶于水中,获得混合溶液;
[0026]
(s2)用强碱性氢氧化物水溶液将步骤(s1)获得的混合溶液的ph值调节至6-7;
[0027]
(s3)将乳化剂溶解于所述步骤(s2)获得的体系中;
[0028]
(s4)向步骤(s3)获得的体系中加入刚性疏水单体并通入惰性气体20min-2h,随后加入引发剂,并继续通入惰性气体10-60min,
[0029]
(s5)将步骤(s4)获得的体系的温度升温至40-70℃并在该温度下反应2-12h,随后降至室温,并纯化1.5-3h,即可获得本发明疏水缔合型阴离子聚丙烯酰胺。
[0030]
在胶束共聚合体系中,水溶性的引发剂引发丙烯酰胺生成大分子链增长自由基,而生成的自由基则在胶束界面引发胶束内的疏水单体进行聚合,小段的疏水段便引入到了亲水聚合物链上,此后大分子自由基离开胶束与丙烯酰胺继续反应,当碰到另一个增溶胶
束后便再次发生反应形成另一个疏水嵌段,最终获得本发明的疏水缔合型阴离子聚丙烯酰胺,其所引入的亲水性单体2-丙烯酰胺-2-甲基丙磺酸,极性强且水溶性好,从而使其在用于水性油墨中能够为水作为分散介质提供很好的基础,且磺酸基的静电排斥力,能够使所获得聚丙烯酰胺不呈现卷曲状态,聚合物分子链更加伸展,疏水缔合作用加强。另外,本发明疏水缔合型阴离子聚丙烯酰胺中引入刚性疏水单体,能够有效提高聚丙烯酰胺的稳定性,使其在高温、高剪切下保持着较高的表观粘度,且对油墨体系没有副作用。另外,本发明制备方法工艺简单,原料成本低,具有较好的工业应用价值。本发明的反应过程如下所示。
[0031][0032]
优选地,在本发明制备方法中,所述步骤(s1)中所获得的混合溶液中丙烯酰胺的质量百分比浓度为5-10%,更优选6-8%;2-丙烯酰胺-2-甲基丙磺酸的质量百分比浓度为2.0-3.0%,更优选2.2-2.5%,基于所述步骤(s1)中所获得的混合溶液的总重量计。
[0033]
优选地,在本发明制备方法中,所述强碱性氢氧化物水溶液为质量百分比浓度为2-4的氢氧化钠水溶液或氢氧化钾水溶液,氢氧化钠或氢氧化钾的碱性比较强,加入量比较少,同时引入的钠离子或钾离子可与体系结合,没有产生新的离子,进而也不会影响所获得产物的性能。
[0034]
优选地,在本发明制备方法中,所述丙烯酰胺、2-丙烯酰胺-2-甲基丙磺酸和刚性疏水单体的摩尔比为(15-35):(4-15):1,进一步优选(15-25):(4-7):1,更优选19:5:1,在该摩尔比范围内,所获得的疏水缔合型阴离子聚丙烯酰胺的流动性和增稠性较好。
[0035]
优选地,在本发明制备方法中,所述乳化剂即为表面活性剂,可为阴离子表面活性剂或阳离子表面活性剂。
[0036]
进一步优选地,在本发明制备方法中,所述阴离子表面活性剂为碳原子数为10-14的烷基硫酸钠如十二烷基硫酸钠、碳原子数为10-14的烷基苯磺酸钠如十二烷基苯磺酸钠中的一种或多种。
[0037]
所述阳离子表面活性剂为碳原子数为10-14的烷基聚氧乙烯醚如十二烷基酚聚氧乙烯醚(op-10)中的至少一种。
[0038]
最优选地,所述乳化剂为十二烷基硫酸钠,这是因为十二烷基硫酸钠的使用相对于反应体系而言不会引入其他离子,进而使所获得产物具有较高的纯度和较少的杂质,且可以合成较高分子量的聚合物。
[0039]
优选地,在本发明制备方法中,所述乳化剂的质量为所述步骤(s1)中所获得的混合溶液的质量的0.5-6.5%,更优选1-6%。
[0040]
优选地,在本发明制备方法中,所述刚性疏水单体为结构式为(ii)的单体:
[0041]
[0042]
变量r1和r2相同或不同且各自独立地为氢、卤素、烷基、取代烷基、芳基、芳烷基、氰基。
[0043]
进一步优选地,在本发明制备方法中,变量r1和r2中的一个代表h,另一个代表卤素、烷基、取代的烷基、芳基、芳烷基、氰基。
[0044]
进一步优选地,在本发明制备方法中,所述烷基为碳原子数为1至51的烷基,更优选碳原子数为1-25的烷基,最优选碳原子数为1-10的烷基如甲基、乙基、丙基、丁基、戊基、己基等等。所述烷基可以为支链的或非支链的。所述烷基若碳原子数过多则会导致单体的疏水性太强,进而导致相应的疏水嵌段的疏水性太强,影响本发明疏水缔合型阴离子聚丙烯酰胺的水溶性。
[0045]
进一步优选地,在本发明制备方法中,所述取代烷基为被以下基团所取代的烷基:卤素、烷基、氰基、苄基、硝基。
[0046]
进一步优选地,在本发明制备方法中,所述芳基为具有单环的碳原子数为6-14个的芳香族碳环基团如苯基,进一步优选地,所述芳基可以为取代的或未取代的芳基,最优选的,所述芳基为苯基或者被卤素取代的苯基。这些基团进行取代后,含取代基的刚性疏水嵌段的空间位阻更大,极性更强,有助于增强本发明疏水缔合型阴离子聚丙烯酰胺的水溶性。
[0047]
进一步优选地,在本发明制备方法中,所述芳烷基是指式-烷基-芳基的基团,其中烷基和芳基是按上面定义的。所述芳烷基可以是取代的或未取代的,其中取代的芳烷基可以在该基团的烷基、芳基或者烷基和芳基部分处被取代。代表性的芳烷基包括但不限于苄基和苯乙基。
[0048]
进一步优选地,在本发明制备方法中,所述卤素为氟、氯、溴或者碘。卤素作为憎水基团,通过其取代可以进一步增加本发明疏水缔合型阴离子聚丙烯酰胺的疏水性,特别是f原子的取代,可以降低表面能,可以很好地提高本发明疏水缔合型阴离子聚丙烯酰胺的疏水性
[0049]
特别需要说明的是,所述刚性疏水单体与本发明疏水缔合型聚丙烯酰胺中的聚合度为c的嵌段一致,故而刚性疏水单体中的变量也与聚合度为c的嵌段中的基团变量一致。
[0050]
优选地,在本发明制备方法中,所述引发剂为过硫酸盐(如过硫酸铵、过硫酸钾、过硫酸铵)或氧化-还原引发剂(如过硫酸铵-亚硫酸钠、过硫酸铵-亚硫酸氢钠、过硫酸钾-亚硫酸钠、过硫酸钾-亚硫酸氢钠、过硫酸铵-硫代硫酸钠或过硫酸钾-硫代硫酸钠)。
[0051]
优选地,在本发明制备方法中,所述引发剂的摩尔量为所述丙烯酰胺、2-丙烯酰胺-2-甲基丙磺酸和刚性疏水单体的摩尔总量的0.1-0.5%。
[0052]
优选地,在本发明制备方法中,所述惰性气体为氮气和/或氦气,基于成本考虑,更优选氮气。在本发明制备方法中通入氮气是为了去除体系中存在的氧气以免引发剂被氧化,从而失去活性,同时也避免由于氧气的存在而影响产物的色泽而使产物颜色偏暗黄。
[0053]
优选地,在本发明制备方法中,所述纯化是指利用甲醇进行纯化以将未反应的单体及乳化剂进行去除,纯化操作可重复数次如2-3次,然后在如45-60℃的温度下进行烘干。
[0054]
另外,需要注意的是,在本发明中所用的水均为去离子水,以免引入不必要的杂质影响最终反应产物的性能。
[0055]
在本发明的第三方面,本发明提供上述疏水缔合型阴离子聚丙烯酰胺的用途,所述用途为所述疏水缔合型阴离子聚丙烯酰胺作为增稠剂应用于水性油墨中,其中所述疏水
缔合型阴离子聚丙烯酰胺的质量占所述水性油墨的总质量的0.5-1%。
[0056]
本发明中制备的疏水缔合型阴离子聚丙烯酰胺首次应用于水性油墨体系中,其与缔合型聚氨酯和缔合型聚丙烯酸相比,疏水缔合型阴离子聚丙烯酰胺增稠剂在制备工艺上可以调控疏水基的微嵌段长度,同时可以降低工艺和原料成本,能够弥补这两者在水性涂料中的的不足。
[0057]
与现有技术相比,本发明具有以下有益效果。
[0058]
(1)本发明疏水缔合型阴离子聚丙烯酰胺引入了刚性疏水单体,能够有效提高聚合物的化学稳定性,使其在高温、高剪切下保持较高的表观粘度,进而将其应用于水性油墨体系中可以提高水性油墨在生产、储运和印刷过程的综合稳定性。
[0059]
(2)本发明的疏水缔合型聚丙烯酰胺引入2-丙烯酰胺-2-甲基丙磺酸嵌段极性强且水溶性好,从而使其在用于水性油墨中能够为水作为分散介质提供很好的基础,且磺酸基的静电排斥力,能够使所获得聚丙烯酰胺不呈现卷曲状态,聚合物分子链更加伸展,疏水缔合作用加强,进而在其作为水性油墨增稠剂使用时具有优异的增稠性并提高水性油墨的稳定性。
[0060]
(3)本发明疏水缔合型阴离子聚丙烯酰胺的制备方法工艺简单,原料成本低,具有较好的工业应用价值,为大规模使用提供必要的基础。
附图说明
[0061]
图1为本发明制备实施例1制备的疏水缔合型阴离子聚丙烯酰胺的dsc谱图。
[0062]
图2为本发明制备实施例5制备的疏水缔合型阴离子聚丙烯酰胺的荧光光谱图。
[0063]
图3为本发明制备实施例5制备的疏水缔合型阴离子聚丙烯酰胺溶液不同浓度与i1/i3的峰强度比的关系图。
[0064]
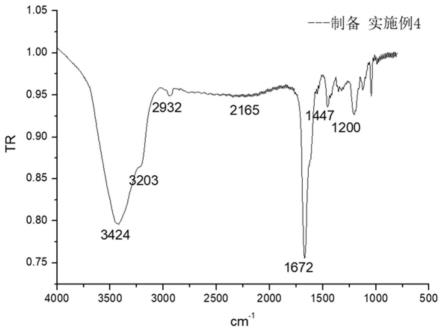
图4为本发明制备实施例4制备的疏水缔合型阴离子聚丙烯酰胺的红外谱图。
[0065]
图5为本发明制备实施例1制备的疏水缔合型阴离子聚丙烯酰胺的紫外谱图。
具体实施方式
[0066]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将以具体实施例和附图详细介绍本发明的技术方案。
[0067]
以下实施例中,所采用的水均为去离子水以避免引入不必要的杂质。所述丙烯酰胺、2-丙烯酰胺-2-甲基丙磺酸、刚性疏水单体、引发剂和乳化剂均为分析纯,其购自国药集团化学试剂有限公司。商业水性油墨增稠剂分别采用的是购自美国陶氏化学公司的罗门哈斯亚乐顺rm-8w和罗门哈斯亚乐顺ase-60,分别为疏水缔合型聚氨酯和疏水缔合型聚丙烯酸。所采用的氢氧化钠溶液的质量百分比浓度为3%。
[0068]
制备实施例
[0069]
制备实施例1
[0070]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和98.64g水于四口烧瓶中配成水溶液,获得混合溶液;
[0071]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0072]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的
调节ph后的体系中;
[0073]
(s4)向步骤(s3)获得的体系中加入0.4g苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0074]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a1。
[0075]
制备实施例2
[0076]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和98.64g水于四口烧瓶中配成水溶液,获得混合溶液;
[0077]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0078]
(s3)在搅拌条件下将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0079]
(s4)向步骤(s3)获得的体系中加入0.48g 4-氟苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0080]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a2。
[0081]
制备实施例3
[0082]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和98.93g水于四口烧瓶中配成水溶液,获得混合溶液;
[0083]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0084]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0085]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0086]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化2次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a3。
[0087]
制备实施例4
[0088]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和100.2g水于四口烧瓶中配成水溶液,获得混合溶液;
[0089]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7;
[0090]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0091]
(s4)向步骤(s3)获得的体系中加入0.62g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气20分钟;
[0092]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a4。
[0093]
制备实施例5
[0094]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和110g水于四口烧瓶中配成水溶液,获得混合溶液;
[0095]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0096]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0097]
(s4)向步骤(s3)获得的体系中加入0.74g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气25分钟;
[0098]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化3次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a5。
[0099]
制备实施例6
[0100]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和97g水于四口烧瓶中配成水溶液,获得混合溶液;
[0101]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0102]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0103]
(s4)向步骤(s3)获得的体系中加入0.3g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0104]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a6。
[0105]
制备实施例7
[0106]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和99.42g于四口烧瓶中配成水溶液,获得混合溶液;
[0107]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0108]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0109]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0110]
(s5)将步骤(s4)获得的体系的温度升温至60℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a7。
[0111]
制备实施例8
[0112]
(s1)8g丙烯酰胺、3g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和116g水于四口烧瓶中配成水溶液,获得混合溶液;
[0113]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0114]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0115]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0116]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应7h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化3次,随后在50℃下烘干,即可获得本发明疏水缔合阴离子聚丙烯酰胺,记作a8。
[0117]
对比实施例
[0118]
对比实施例1
[0119]
(s1)8g丙烯酰胺、2.58g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和94.12g水于四口烧瓶中配成水溶液,获得混合溶液;
[0120]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0121]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0122]
(s4)向步骤(s3)获得的体系中加入0.035g过硫酸钾引发剂,并通入氮气30分钟;
[0123]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得产物,记作b1。
[0124]
对比实施例2
[0125]
(s1)8g丙烯酰胺和78g水于四口烧瓶中配成水溶液,获得混合溶液;
[0126]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0127]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0128]
(s4)向步骤(s3)获得的体系中加入0.46g苯乙烯(单体摩尔总量的3%)并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0129]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得产物,记作b2。
[0130]
对比实施例3
[0131]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和95g水于四口烧瓶中配成水溶液,获得混合溶液;
[0132]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0133]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0134]
(s4)向步骤(s3)获得的体系中加入0.09g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0135]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b3。
[0136]
对比实施例4
[0137]
(s1)8g丙烯酰胺和2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和110g水于四口烧
瓶中配成水溶液,获得混合溶液;
[0138]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0139]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0140]
(s4)向步骤(s3)获得的体系中加入1.48g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0141]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化100min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b4。
[0142]
对比实施例5
[0143]
(s1)8g丙烯酰胺、1.18g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和85.2g水于四口烧瓶中配成水溶液,获得混合溶液;
[0144]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0145]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0146]
(s4)向步骤(s3)获得的体系中加入0.42g4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0147]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b5。
[0148]
对比实施例6
[0149]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和116g水于四口烧瓶中配成水溶液,获得混合溶液;
[0150]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0151]
(s3)在搅拌条件下,将8.89g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0152]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0153]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b6。
[0154]
对比实施例7
[0155]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和98.4g水于四口烧瓶中配成水溶液,获得混合溶液;
[0156]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至8,
[0157]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0158]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0159]
(s5)将步骤(s4)获得的体系的温度升温至50℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b7。
[0160]
对比实施例8
[0161]
(s1)8g丙烯酰胺、2.66g 2-丙烯酰胺基-2-甲基-1-丙烷磺酸和98.4g水于四口烧瓶中配成水溶液,获得混合溶液;
[0162]
(s2)用氢氧化钠溶液将步骤(s1)获得的混合溶液的ph值调节至7,
[0163]
(s3)在搅拌条件下,将1.11g乳化剂十二烷基硫酸钠溶解于所述步骤(s2)获得的调节ph后的体系中;
[0164]
(s4)向步骤(s3)获得的体系中加入0.46g 4-甲基苯乙烯并向体系中通入氮气30min,随后加入0.035g过硫酸钾引发剂,并通入氮气10分钟;
[0165]
(s5)将步骤(s4)获得的体系的温度升温至80℃并在该温度下反应6h,反应完毕后,降低至室温并利用甲醇纯化120min并反复纯化2次,随后在50℃下烘干,即可获得疏水缔合阴离子聚丙烯酰胺,记作b8。
[0166]
测试实施例
[0167]
测试实施例1表征测试
[0168]
将本发明制备实施例1制备的疏水缔合阴离子聚丙烯酰胺以下性能表征,测试结果示于附图1-4中。
[0169]
dsc测试:采用的仪器是购自梅特勒-托利多集团的tga/dsc,首先在25-400℃下,加热速率10℃/min,n
2 100ml/min的条件下做空白样的测试,然后称取适量的样品在25-400℃下,加热速率10℃/min,n
2 100ml/min条件下测试。
[0170]
荧光谱图测试:采用fls1000荧光光谱仪,芘的最终浓度是5*10-6
mol/l,测试温度25℃,荧光激发波长335nm,激发和发射狭缝2.5nm,扫描范围350-550nm,样品池厚度1cm。
[0171]
红外谱图测试:采用的仪器是bruker tensorⅱ,测试的波长是400-4000nm。
[0172]
紫外谱图测试:采用的仪器是lambda 850+紫外分光光度计,测试的波长是200-350nm。
[0173]
通过对图1分析可发现,在85℃以及322℃附近分别存在一个玻璃化转变温度,322℃处对应的玻璃化转变温度为分子链中丙烯酰胺和2-丙烯酰胺-2-甲基丙磺酸(amps)嵌段共聚链段中的tg,明显高于丙烯酰胺均聚物的tg(163℃),这是因为分子链中的amps单元的极性以及庞大的侧基增大的空间位阻,增强了分子链的极性,提高了分子链的刚性,使得共聚物分子链内旋转困难,使得聚合物的亲水链段tg较高,85℃附近对应于疏水链段的tg,表明该聚合物为嵌段共聚物,并且刚性疏水单体以微嵌段的形式分布于分子链上,因为合成的试样完全溶于水,说明聚合物中不可能有疏水单体均聚物存在。
[0174]
对图2和图3进行分析可发现,芘探针的荧光发射光谱依次在373nm、379nm、384nm、390nm和410nm附近出现五重发射峰,且i1/i3值随聚合物浓度的增加而减少。低浓度时(<0.5g/l),i1/i3值较大,且随浓度的变化幅度较小,反映了芘探针所处环境的极性较大。同时,由于在低浓度下,大分子之间的距离较远,聚合物分子的疏水基团之间发生疏水缔合的机率和数量均很小,即使存在分子内缔合,也会因聚合物浓度太低而使得疏水微区的量很少,且磺酸基团间的静电斥力使聚合物大分子链呈疏松伸展状态。而当聚合物浓度达到
0.5g/l时,i1/i3开始缓慢下降,此时溶液中以分子内缔合为主,此后,i1/i3值急剧下降,表明溶液中生成了大量疏水微区,可见本发明合成疏水缔合型阴离子聚丙烯酰胺具有很好的疏水缔合效果,可以起到很好的增稠效果。
[0175]
通过图4并经分析可知,3424cm-1对应酰胺的n-h伸缩振动;3203cm-1对应于苯环上c-h伸缩振动;2932cm-1对应于-ch3的伸缩振动,1447cm-1对应苯环c=c双键振动吸收峰;1672cm-1对应于酰胺c=o伸缩振动;1015cm-1和1043cm-1对应于so
3-的振动,表明本发明制备实施例1中的制备过程将丙烯酰胺、2-丙烯酰胺基-2-甲基-1-丙烷磺酸、苯乙烯进行了有效的聚合,得到了疏水缔合阴离子聚丙烯酰胺。
[0176]
通过图5可以发现,在212nm处存在明显的吸收峰,对应于聚合物主链上的苯环结构,而苯乙烯中苯环的吸收峰在225nm处,出现了一定程度的红移,这主要归结于苯乙烯中乙烯基双键的存在,增大了苯环的共轭体系,使得价电子跃迁所需要的能量变低,发生很大程度地红移,进一步表明刚性疏水单体嵌段到分子链上。
[0177]
另外,本发明采用gb12005.1-89“聚丙烯酰胺特性粘数测定方法”中规定的测试方法测定粘均分子量,其采用氯化钠溶液做溶剂,并制备一定浓度的试样溶液,使用气承液柱式乌式粘度计,分别测定溶液和溶剂的流经时间,根据测定的值计算特性粘数,进而算出粘均分子量,上述制备实施例中制备的粘均分子量在8*105~1.7*106之间。
[0178]
测试实施例2稳定性测试
[0179]
将发明实施例1-8制备的疏水缔合阴离子聚丙烯酰胺a1-a8、对比实施例1-8制备的产物b1-b8、市售疏水缔合聚丙烯酸增稠剂、市售疏水缔合聚氨酯增稠剂、自制水性油墨(不含增稠剂)进行稳定性测试,具体测试过程如下,并将结果示于下表1中。
[0180]
自制油墨:按照基于所述水性油墨的总重量计重量百分数分别为以下进行各组分配制水性油墨:30%水性树脂、10%有机颜料、0.5%增稠剂、0.2%氨水、0.3%消泡剂和59%水,然后将水性树脂、颜料、增稠剂、消泡剂、水加入容器中以500r/min的条件搅拌30分钟,搅拌过程中滴加氨水,然后加入与体系相同质量的研磨珠在1500r/min下搅拌打磨1h,得到水性油墨。
[0181]
稳定性测试:将自制油墨与增稠剂混合均匀并放置在50℃的环境中静置观察,通过开始沉降时间来测试油墨稳定性性能,期所用增稠剂的质量基于所述自制油墨和所述增稠剂的总质量的0.5%。
[0182]
表1水性油墨稳定性测试结果
[0183]
测试项目稳定时间(h)测试项目稳定时间(h)a148b11a253b22a360b340a455b445a550b550a658b658a762b759a853b861聚丙烯酸25聚氨酯24
未加入增稠剂水性油墨0.5
‑‑‑‑
[0184]
通过对上述实施例的数据进行分析发现,烷基取代的疏水单体制备的增稠剂,在水性油墨体系中更有助于水性油墨的稳定性(即制备实施例1、2、3),能有效延长其保质期,疏水单体通过微嵌段的形成链接到分子链上,更有利于聚合物溶液分子间缔合,随着疏水单体的摩尔比的增加(即制备实施例4、5、6),制备的增稠剂在油墨配方中的稳定性存在一个先升高后降低的趋势,这主要在于疏水含量过高导致疏水链段过长,疏水缔合作用发生在分子内,易出现分子链卷曲,不利于分子间疏水缔合。增稠剂的合成环境在中性或者弱酸性,如果在碱性环境下合成的聚合物,聚合物容易分解,影响其增稠效果(即制备实施例3和对比实施例7)。另外,合适的温度更有利于聚合物的合成,温度过高,会导致聚合速率难以控制,发生爆聚现象(即制备实施例7、8和对比实施例8)。同时亲水单体amps的so
3-离子的静电排斥作用有助于分子间的疏水缔合,但是过多的-so
3-的静电排斥反而会削弱分子间缔合(即对比实施例5和6);乳化剂的用量会影响疏水缔合作用,乳化剂的含量增加,当高于6%时,溶解有刚性疏水单体的胶束数量大,每个胶束中的刚性疏水分子数减少,导致疏水链段较短,使得疏水缔合作用减弱(即制备实施例3和对比实施例6)。
[0185]
根据上述说明书的揭示和教导,本发明所属领域的技术人员还可以对上述实施方式进行变更和修改。因此,本发明并不局限于上面揭示和描述的具体实施方式,对发明的一些修改和变更也应当落入本发明的权利要求的保护范围内。此外,尽管本说明书中使用了一些特定的术语,但这些术语只是为了方便说明,并不对本发明构成任何限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1