一种高纯度地诺前列腺素的制备方法与流程
1.本发明涉及药物化学生产技术领域,具体涉及一种高纯度地诺前列腺素的制备方法。
背景技术:
2.前列腺素(prostaglandins,简称pgs)是一类具有广泛生理活性的重要内源性产物,存在于几乎所有哺乳动物组织中,在生殖、消化、呼吸和心血管系统中发挥着重要作用,参与体温调节、炎症反应、青光眼、妊娠、高血压、溃疡及哮喘等生理病理过程。
3.org.biomol.chem.2017,15,6281
–
6301描述了pgs结构特征:具有一个五元脂环及两个侧链,上侧链α通常有7个碳酸、下侧链ω有8个碳组成20碳不饱和脂肪酸及其类似物,结构式表示如下:
[0004][0005]
pgs最早由美国学者von eluer在1930年发现并命名,1962年bergstorm提取出两种pg纯品(pgfl和pgf2)并确定其化学结构;1969年willis首次提出pgs是体内一种炎症介质后,相关各种生理和药理活性得以深入研究。
[0006]
前列腺素天然来源少,提取困难,体内代谢迅速,稳定性差等缺点,科学家基于天然前列腺素的高活性,结构新颖性以及其不稳定性等特点,相继改造并合成了一系列前列腺素类似物,以满足临床需求。nat.chem.2021,13,692
–
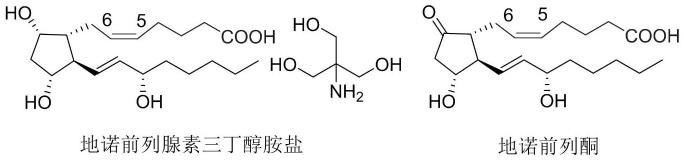
697报道截止2019末为止,超过20种前列腺类似物进入市场,其中不乏超过十亿美圆的比马前列素bimatoprost,充分显示其在制药工业的重要性和独特价值。比较早期就进入临床,应用于引产、催生和人工流产前列腺素药物有:地诺前列腺素和卡地诺前列酮等。
[0007][0008]
目前根据现有文献调研,制备地诺前列腺素氨基丁三醇f2a的路线主要是以下两种策略:a、先合成带有任意一条侧链的五元环结构,再通过1,4-加成引入另一侧链,该合成方法收率高,但不可避免上侧链因wittig偶联反应而产生5,6位双键3~8%反式异构体,从而影响最终产物的质量和收率;b、三组分偶联法,即“一锅法”同时引入上侧链和下侧链,这个方法虽然路线短,原料廉价易得,成本优势明显,但是这个方法关键步骤副产物多,收率低,产品质量无法保障,因此也无法工业化生产。
技术实现要素:
[0009]
为了克服上述技术缺陷,本发明提供了一种高纯度地诺前列腺素的制备方法。利用大环内酯2作为起始物料,合成高质量的地诺前列腺素,该方法不仅成功避免了地诺前列腺5,6位双键反式异构体产生,而且路线更短,收率更好,质量更为可控。
[0010]
本发明高纯度地诺前列腺素的制备方法,反应路线表示如下:
[0011][0012]
包括如下步骤:以科里大环内酯2为起始原料,与磷脂试剂3进行wittig-horner反应,得到烯酮中间体4,接着在手性还原剂存在下不对称还原反应,得到15位s构型中间体5,脱硅保护基后得到双醇中间体6,重结晶除去15r异构体,水解后得到羧酸中间体7,最后和三丁醇胺成盐,得到地诺前列腺素氨基丁三醇8。
[0013]
进一步地,在上述技术方案中,所述科里大环内酯2采用化合物1按照文献方法制备。参考cn115010692a:
[0014]
进一步地,在上述技术方案中,所述wittig-horner反应为采用nah、n-buli或lda与磷脂试剂3去质子后,再与科里大环内酯2反应。
[0015]
进一步地,在上述技术方案中,所述wittig-horner反应,磷脂试剂3与大环内酯2摩尔比为1.5:1,反应温度为0~5℃。
[0016]
进一步地,在上述技术方案中,所述手性还原剂为r-mecbs,与bh3-me2s或bh3-thf组合进行不对称还原。
[0017]
进一步地,在上述技术方案中,所述不对称还原时,r-mecbs与中间体4摩尔比为1:10,反应温度为-20~0℃,反应溶剂为四氢呋喃或二氯甲烷。
[0018]
进一步地,在上述技术方案中,所述脱thp保护在ppts、三氟乙酸或盐酸催化剂存在下进行。
[0019]
进一步地,在上述技术方案中,所述脱thp保护时,催化剂与中间体5摩尔比为0.1-0.2:1。
[0020]
进一步地,在上述技术方案中,所述重结晶采用乙酸乙酯和石油醚混合溶剂。
[0021]
进一步地,在上述技术方案中,所述重结晶时,乙酸乙酯与石油醚体积比为1:2-5。
[0022]
进一步地,在上述技术方案中,所述水解反应采用氢氧化钠、氢氧化钾或氢氧化锂。
[0023]
进一步地,在上述技术方案中,所述水解时,碱与中间体6摩尔比为1-5:1。
[0024]
发明有益效果
[0025]
本发明利用科里大环内酯作为起始物料来制备地诺前列腺素,从源头革除了传统wittig反应而不可避免产生上侧链5,6-反式双键异构体的难题;同时利用不对称催化还原,主要得到15s中间体,通过重结晶,将15r异构体彻底去除,从而使得该产品质量大大提高,增强了产品的竞争力。
具体实施方式
[0026]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。本发明所用试剂和原料均市售可得。
[0027]
实施例1:起始物料大环内酯醛2的制备
[0028]
详细操作参考cn115010692a。
[0029]
实施例2:中间体4的制备
[0030]
氮气保护下,在100ml四氢呋喃溶液中,加入60%nah(1.8g,45.0mmol),冷却到0~5℃滴加市售磷脂3(10.0g,45.0mmol),加毕搅拌反应30分钟,向其中滴加大环内酯醛2(10.0g,28.4mmol)。加毕继续反应,直至起始物料大环内酯醛2完全消失。常规后处理,柱层析得到9.8g烯酮中间体4,无色油状液体,收率77%。1h-nmr(400mhz,cdcl3):6.61(dd,j=9.2,15.6hz,1h),6.24(d,j=9.2hz,1h),5.34(m,1h),5.23(m,2h),3.91(dd,j=8.0,16.8hz,1h),2.60-2.37(m,7h),2.24(m,1h),2.10(m,2h),1.90-1.80(m,3h),1.39-1.21(m,4h),0.90(t,j=6.8hz,3h),0.85(s,9h),0.00(s,6h).
[0031]
实施例3:化合物5的制备
[0032]
氮气保护下,向100ml干燥三口瓶中,加入1m硼烷四氢呋喃溶液(30ml,30.0mmol),冷却到0~5℃,向其中加入还原剂r-2-甲基-cbs-恶唑硼烷(0.83g,3.0mmol),继续在该温度下搅拌1小时,冷却至-20~-30℃,向其中滴加实施例2中得到的烯酮中间体4(9.0g,21.4mmol),加毕,tlc跟踪至原料完毕完毕,停止反应。常规后处理,柱层析得到8.0g手性烯丙醇中间体5,无色油状液体,收率89%。
[0033]
实施例4:化合物6的制备
[0034]
氮气保护下,在100ml三口瓶中,加入实施例3中得到的烯丙醇中间体5(8.0g,19.0mmol)和甲醇(50ml),搅拌下加入ppts(0.8g,3.18mmol),然后升温回流,反应3~5小时,tlc检测原料消失,停止反应。减压浓缩至干,向其中加入100ml乙酸乙酯溶解,水和饱和食盐水洗,无水硫酸钠干燥,过滤浓缩至干,乙酸乙酯和石油醚重(体积比1/2)结晶,干燥至恒重,得到5.5g双醇中间体6,白色固体,收率为86%。1h-nmr(400mhz,cdcl3):5.59(dd,j=7.2,15.2hz,1h),5.38(dd,j=8.8,15.2hz,1h),5.30(m,1h),5.15(m,2h),4.04(dd,j=8.0,13.6hz,1h),3.80(dd,j=8.0,16.0hz,1h),2.50(m,1h),2.24(m,1h),2.40-1.20(m,18h),0.81(t,j=6.8hz,3h).lc-ms:(m/z):359.2[m+na]
+
。
[0035]
实施例5:化合物7制备
[0036]
氮气保护下,将双醇中间体6(5.0g,14.8mmol)溶解于50ml甲醇中,冷却到0~10℃,向其中加入10%氢氧化锂水溶液(40ml),然后慢慢升至室温,直至原料完全消失为止。浓缩至干,加入1m盐酸调ph=4-5,乙酸乙酯萃取三次,合并有机相,浓缩至干,得到5.27g油状物羧酸中间体7,收率定量,未经纯化直接进行下一步反应。
[0037]
实施例6:地诺前列腺素8的制备
[0038]
将实施例5中得到的羧酸中间体7(5.27g,14.8mmol)溶解于50ml丙酮,然后加入三丁醇胺(1.80g,14.9mmol)溶解于5ml水中,然后加热至40~50℃,反应5小时,浓缩至干,加入丙酮100ml,室温搅拌10~20小时,析出固体,过滤,真空干燥,得到5.8g地诺前列腺素8,白色固体,收率82%。1h-nmr(400mhz,d2o):5.47-5.35(m,4h),4.10(m,1h),4.02(dd,j=6.4,13.2hz,1h),3.80(dd,j=6.4,16.0hz,1h),3.78(s,6h),2.43(m,1h),2.34(m,1h),2.15-1.93(m,6h),1.54-1.24(m,6h),1.20(m,6h),0.76(t,j=6.8hz,3h).lc-ms:(m/z):377.3[m+na]
+
。
[0039]
实施例7中间体4的制备
[0040]
氮气保护下,在100ml四氢呋喃溶液中,加入60%nah(3.6g,90.0mmol),冷却到0~5℃滴加市售磷脂3(20.0g,90.0mmol),加毕搅拌反应30分钟,向其中滴加大环内酯醛2(10.0g,28.4mmol)。加毕继续反应,直至起始物料大环内酯醛2完全消失。常规后处理,柱层析得到9.0g烯酮中间体4,无色油状液体,收率71%。
[0041]
实施例8中间体4的制备
[0042]
氮气保护下,在100ml四氢呋喃溶液中,加入市售磷脂3(10.0g,45.0mmol),搅拌下,完全溶解,冷却至0~5℃,然后向其中滴加lda(1.5m,30ml,45.0mmol),加毕,搅拌反应30分钟,向其中滴加大环内酯醛2(10.0g,28.4mmol)。加毕继续反应,直至起始物料大环内酯醛2完全消失。常规后处理,柱层析得到8.1g烯酮中间体4,无色油状液体,收率64%。
[0043]
实施例9中间体4的制备
[0044]
氮气保护下,在100ml四氢呋喃溶液中,加入市售磷脂3(10.0g,45.0mmol),搅拌下,完全溶解,冷却至0~5℃,然后向其中滴加lda(1.5m,30ml,45.0mmol),加毕,搅拌反应30分钟,向其中滴加大环内酯醛2(10.0g,28.4mmol)。加毕继续反应,直至起始物料大环内酯醛2完全消失。常规后处理,柱层析得到8.1g烯酮中间体4,无色油状液体,收率64%。
[0045]
实施例10:化合物5的制备
[0046]
氮气保护下,向100ml干燥三口瓶中,加入1m硼烷四氢呋喃溶液(30ml,30.0mmol),冷却到0~5℃,向其中加入还原剂r-2-甲基-cbs-恶唑硼烷(0.59g,2.14mmol),继续在该温度下搅拌1小时,冷却至-20~-30℃,向其中滴加实施例2中得到的烯酮中间体4(9.0g,21.4mmol),加毕,tlc跟踪至原料完毕完毕,停止反应。常规后处理,柱层析得到7.2g手性烯丙醇中间体5,无色油状液体,收率80%。
[0047]
实施例11:化合物5的制备
[0048]
氮气保护下,向100ml干燥三口瓶中,加入1m硼烷四氢呋喃溶液(30ml,30.0mmol),冷却到0~5℃,向其中加入还原剂r-2-甲基-cbs-恶唑硼烷(1.8g,6.5mmol),继续在该温度下搅拌1小时,冷却至-20~-30℃,向其中滴加实施例2中得到的烯酮中间体4(9.0g,21.4mmol),加毕,tlc跟踪至原料完毕完毕,停止反应。常规后处理,柱层析得到8.1g手性烯丙醇中间体5,无色油状液体,收率89%。
[0049]
实施例12:化合物6的制备
[0050]
氮气保护下,在100ml三口瓶中,加入烯丙醇中间体5(8.0g,19.0mmol)和甲醇(50ml),搅拌下加入对甲苯磺酸(0.5g),然后升温回流,反应3~5小时,tlc检测原料消失,停止反应。减压浓缩至干,向其中加入100ml乙酸乙酯溶解,水和饱和食盐水洗,无水硫酸钠干燥,过滤浓缩至干,乙酸乙酯和石油醚重结晶,干燥至恒重,得到5.0g双醇中间体6,白色固体,收率为78%。
[0051]
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1