一种改性环氧树脂及其制备方法、UV环氧树脂灌封胶与流程
一种改性环氧树脂及其制备方法、uv环氧树脂灌封胶
技术领域
1.本发明属于环氧树脂技术领域,具体涉及一种改性环氧树脂及其制备方法、uv环氧树脂灌封胶。
背景技术:
2.灌封就是将液态胶水用机械或手工方式灌入装有电子元件、线路的器件内,在常温或加热条件下固化成为性能优异的热固性高分子绝缘材料。其中,uv环氧树脂是常用的灌封胶。
3.灌封胶不仅要具有颜色透明、黏度低、固化收缩小、膨胀系数小、耐高低温交变性能好等特性,而且还必须具有一些特定的功能,如韧性高、固化时间短、固化深度高、抗指划痕能力强。目前对uv环氧树脂增韧方法通常有3种,第1种是在环氧分子或固化剂分子中引入柔性链链段,第2种是在环氧树脂配方中添加一定量的活性增柔剂或增韧剂,第3种是在配方中加入改性的硅粉。
4.但是以上3种方法在增韧时,很难同时满足固化时间、固化深度和抗指划痕性能。
技术实现要素:
5.本发明的目的在于提供一种改性环氧树脂及其制备方法、uv环氧树脂灌封胶,利用本发明改性环氧树脂制备的uv环氧树脂灌封胶韧性高且固化时间短、固化深度高、抗指划痕能力强。
6.本发明提供了一种改性环氧树脂,包括环氧树脂和接枝于所述环氧树脂上的非活性纳米粒子;所述环氧树脂与所述非活性纳米粒子通过共价键相连;所述非活性纳米粒子包括非活性纳米硅粉、非活性纳米埃洛石、非活性纳米二氧化钛和非活性纳米白炭黑中的一种;
7.所述共价键由所述环氧树脂重复单元中的羟基与所述非活性纳米粒子的羟基进行缩合反应生成;
8.所述环氧树脂重复单元组成的聚合链段中羟基呈等间距分布。
9.优选的,所述环氧树脂包括双酚a环氧树脂、双酚f环氧树脂、双酚s环氧树脂和氢化双酚a型环氧树脂中的一种或多种。
10.优选的,还包括硅烷偶联剂,所述环氧树脂与所述非活性纳米粒子还通过硅烷偶联剂相连。
11.优选的,所述硅烷偶联剂包括γ-氨丙基三乙氧基硅烷、γ-环氧丙氧基丙基三甲氧基硅烷和苯胺甲基三甲氧基硅烷中的一种。
12.本发明还提供了上述方案所述改性环氧树脂的制备方法,包括以下步骤:
13.将非活性纳米粒子分批加入到将环氧树脂中,进行缩合反应,得到所述改性环氧树脂。
14.优选的,当所述环氧树脂与所述非活性纳米粒子还通过硅烷偶联剂相连时,所述
改性环氧树脂的制备方法包括以下步骤:将非活性纳米粒子分批加入到将环氧树脂后,再向所得混合物中加入硅烷偶联剂进行缩合反应,得到所述改性环氧树脂。
15.优选的,所述环氧树脂与所述非活性纳米粒子的质量比为1:0.01~0.15。
16.优选的,所述环氧树脂与所述硅烷偶联剂的质量比为2500~4200:1。
17.优选的,所述缩合反应分为第一缩合反应和第二缩合反应;所述第一缩合反应的温度为110~130℃,时间为1~3h;所述第二缩合反应的温度为室温,时间为8~16h。
18.本发明还提供了一种uv环氧树脂灌封胶,以质量分数计,包括以下组分:
[0019][0020]
改性环氧树脂余量;
[0021]
所述改性环氧树脂为上述方案所述的改性环氧树脂或上述方案所述制备方法制备的改性环氧树脂。
[0022]
本发明提供了一种改性环氧树脂,包括环氧树脂和接枝于所述环氧树脂上的非活性纳米粒子;所述环氧树脂与所述非活性纳米粒子通过共价键相连;所述非活性纳米粒子包括非活性纳米硅粉、非活性纳米埃洛石、非活性纳米二氧化钛和非活性纳米白炭黑中的一种;所述共价键由所述环氧树脂重复单元中的羟基与所述非活性纳米粒子的羟基进行缩合反应生成;所述环氧树脂重复单元组成的聚合链段中羟基呈等间距分布。环氧树脂的重复单元组成的聚合链段中羟基呈等间距分布,具有规律性。非活性纳米粒子通过其表面的羟基与环氧树脂中规律性的羟基通过缩合反应规律性地修饰在环氧树脂表面,改善了非活性纳米粒子在uv环氧树脂灌封胶中的分散性,减少了非活性纳米粒子的团聚,并且使环氧树脂的空间更加紧凑,大大延长了体系的沉降时间;在uv光照射时,规律性修饰在环氧树脂的非活性纳米粒子形成了uv光固化通道,集中了更多的光的能量,从而在固化时增加了环氧树脂分子的碰撞机会,减少了光固化时间,增加了环氧树脂的交联密度,增加了光固化深度,进而有效的提高环氧树脂固化后的硬度和固化后的抗指划痕性能;此外,规律性修饰在环氧树脂上的非活性纳米粒子和环氧树脂形成的具有强相互作用的化学键也提升了固化后灌封胶的韧性。实施例结果表明,本发明改性环氧树脂制备的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为85~90s,固化深度0.20~0.30mm,邵氏硬度为85~90;拉伸强度为66.40mpa;弯曲强度为133.2mpa;冲击强度为13.40kj/mm2;固化后无表面指划痕;对载带的粘接性失效模式良好,弯折测试表现韧性较高。
[0023]
进一步地,本发明中的硅烷偶联剂使环氧树脂结构单元上的羟基反应完全。
附图说明
[0024]
图1为本发明的改性环氧树脂的结构示意图;
[0025]
图2为对比例1的uv环氧树脂灌封胶断面图;
[0026]
图3为对比例2的uv环氧树脂灌封胶断面图;
[0027]
图4为实施例1的uv环氧树脂灌封胶断面图;
[0028]
图5为实施例1中的双酚a环氧树脂和实施例1制得的改性环氧树脂的红外谱图。
具体实施方式
[0029]
本发明提供了一种改性环氧树脂,包括环氧树脂和接枝于所述环氧树脂上的非活性纳米粒子;所述环氧树脂与所述非活性纳米粒子通过共价键相连;所述非活性纳米粒子包括非活性纳米硅粉、非活性纳米埃洛石、非活性纳米二氧化钛和非活性纳米白炭黑中的一种;
[0030]
所述共价键由所述环氧树脂重复单元中的羟基与所述非活性纳米粒子的羟基进行缩合反应生成;
[0031]
所述环氧树脂重复单元组成的聚合链段中羟基呈等间距分布。
[0032]
在本明中,所述环氧树脂优选包括双酚a环氧树脂、双酚f环氧树脂、双酚s环氧树脂和氢化双酚a型环氧树脂中的一种或多种。在本发明中,所述环氧树脂的聚合度优选为30~150,更优选为50~100,进一步优选为60~80。
[0033]
在本发明中,所述双酚a环氧树脂的结构式如式i所示:
[0034][0035]
所述双酚f环氧树脂的结构式如式ii所示:
[0036][0037]
所述双酚s环氧树脂的结构式如式iii所示:
[0038][0039]
在本发明中,所述改性环氧树脂中非活性纳米粒子的接枝量优选为0.1~0.3wt%,更优选为0.2~0.25wt%。
[0040]
在本发明中,所述改性环氧树脂还包括硅烷偶联剂,所述环氧树脂与所述非活性纳米粒子优选还通过硅烷偶联剂相连。在本发明中,所述硅烷偶联剂优选包括γ-氨丙基三乙氧基硅烷、γ-环氧丙氧基丙基三甲氧基硅烷和苯胺甲基三甲氧基硅烷中的一种或多种。本发明中的硅烷偶联剂可以使环氧树脂结构单元上的羟基与非活性纳米粒子反应完全。
[0041]
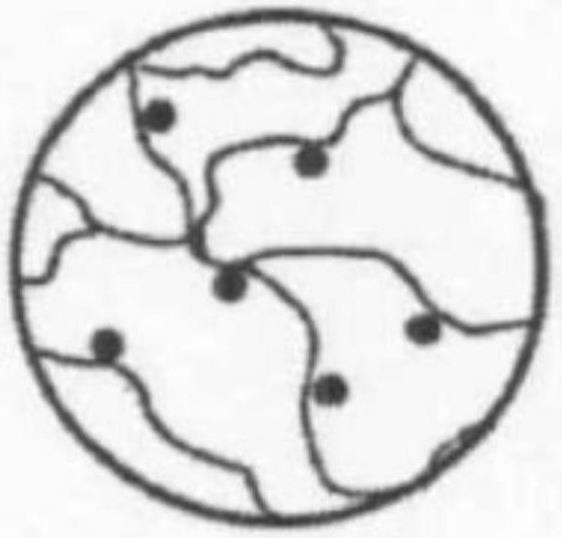
本发明的改性环氧树脂的结构示意图如图1所示,图1中的黑点代表非活性纳米粒子,黑线代表环氧树脂。
[0042]
环氧树脂的重复单元组成的聚合链段中羟基呈等间距分布,具有规律性。非活性纳米粒子通过其表面的羟基与环氧树脂中规律性的羟基通过缩合反应规律性地修饰在环氧树脂表面,改善了非活性纳米粒子在uv环氧树脂灌封胶中的分散性,减少了非活性纳米
粒子的团聚,并且使环氧树脂的空间更加紧凑,大大延长了体系的沉降时间;在uv光照射时,规律性修饰在环氧树脂的非活性纳米粒子形成了uv光固化通道,集中了更多的光的能量,从而在固化时增加了环氧树脂分子的碰撞机会,减少了光固化时间,增加了环氧树脂的交联密度,增加了光固化深度,进而有效的提高环氧树脂固化后的硬度和固化后的抗指划痕性能;此外,规律性修饰在环氧树脂的非活性纳米粒子和环氧树脂形成的具有强相互作用的化学键也提升了固化后灌封胶的韧性。
[0043]
在本发明中,所述非活性纳米粒子指的是在生产过程中,不加入其它改性物质而制备的粒子。
[0044]
本发明还提供了上述所述改性环氧树脂的制备方法,包括以下步骤:
[0045]
将非活性纳米粒子分批加入到环氧树脂中,进行缩合反应,得到所述改性环氧树脂。
[0046]
在本发明中,所述环氧树脂与所述非活性纳米粒子的质量比优选为1:0.01~0.15,更优选为1:0.05~0.12,进一步优选为1:0.06~0.08。在本发明中,所述非活性纳米粒子的粒径优选为0.1~100nm,更优选为10~80nm,进一步优选为30~60nm。在本发明中,所述非活性纳米粒子的制备方法优选为机械球磨法、化学还原法、溶剂热法、气相沉积法和液相急等法中的一种。本发明对于所述机械球磨法、化学还原法、溶剂热法、气相沉积法和液相急等法没有特殊的限定,采用本领域技术人员熟知的方案即可。不同制备方法得到的非活性纳米粒子具有不同的性质,这些不同的性质使得非活性纳米粒子在最终的uv环氧树脂灌封胶中有着细微不同性能方面的表现。例如:机械球磨法制备的非活性纳米硅粉颗粒尺寸分布范围较宽,特别容易分散,最终的uv环氧树脂粘度较低,uv固化后邵氏硬度较低;化学还原法制备的纳米非活性纳米硅粉,制备的纳米硅粉平均尺寸偏大,稳定较好,最终的uv环氧树脂灌封胶防沉降性能较好;溶剂热法制备的非活性纳米硅粉,颗粒度均一,同时可以根据尺寸需要调控制备不同均一尺寸的非活性纳米硅粉,平均颗粒度种类较多,可以根据非活性纳米硅粉尺寸的变化制备出具有不同邵氏硬度和不同固化深度的uv环氧树脂灌封胶;气相沉积法、液相急等法制备的非活性纳米硅粉性能同上述溶剂热法制备的非活性纳米硅粉,制备的uv环氧树脂灌封胶性能也同上。
[0047]
在本发明中,所述环氧树脂的温度优选为70~90℃,更优选为80~85℃。本发明对每批加入非活性纳米粒子的重量以及分批加入的次数没有特殊要求,能够避免非活性纳米粒子团聚即可。在本发明的实施例中,每批加入非活性纳米粒子的重量为总重量的0.5~1%,间隔的时间为3min。本发明通过分批加入可以减少非活性纳米粒子的团聚,提高其分散效果。本发明优选在搅拌条件下将所述非活性纳米粒子加入到环氧树脂中。本发明对所述搅拌的时间和速率没有特殊要求,能够实现混合均匀即可。在本发明的实施例中,具体是在搅拌条件下加入所有非活性纳米粒子后,继续搅拌1h。
[0048]
在本发明中,所述缩合反应优选分为第一缩合反应和第二缩合反应。在本发明中,所述第一缩合反应的温度优选为110~130℃,更优选为120~125℃;时间优选为1~3h,更优选为2~2.5h。所述第二缩合反应的温度优选为室温,时间优选为8~16h,更优选为10~14h,进一步优选为12~13h。在本发明中,所述第一缩合反应和第二缩合反应优选在搅拌的条件下进行,所述搅拌的速度优选独立地为180~220r/min,更优选为200~210r/min。在第一缩合反应过程中,环氧树脂表面的大部分羟基与非活性纳米粒子发生了缩合反应;在第
二缩合反应过程中,环氧树脂中未反应的羟基继续与非活性纳米粒子缩合相接。本发明通过第一缩合反应和第二缩合反应使的环氧树脂中的羟基充分与非活性纳米粒子发生了反应,保证非活性纳米粒子在环氧树脂表面的等间距分布。
[0049]
在本发明中,当所述环氧树脂与所述非活性纳米粒子还通过硅烷偶联剂相连时,所述改性环氧树脂的制备方法优选包括以下步骤:将非活性纳米粒子分批加入到环氧树脂后,再向所得混合物中加入硅烷偶联剂进行缩合反应,得到所述改性环氧树脂。在第一缩合和第二缩合反应过程中,硅烷偶联剂将环氧树脂与非活性纳米粒子相连,使环氧树脂结构单元上的羟基与非活性纳米粒子反应完全。
[0050]
在本发明中,所述环氧树脂与所述硅烷偶联剂的质量比优选为2500~4200:1,更优选为3000~4000:1,进一步优选为3200~3600:1。
[0051]
在本发明中,添加硅烷偶联剂时改性环氧树脂的制备条件同上述不添加硅烷偶联剂时改性环氧树脂的制备条件,这里不再赘述。
[0052]
本发明还提供了一种uv环氧树脂灌封胶,以质量分数计,包括以下组分:
[0053][0054]
改性环氧树脂余量;
[0055]
所述改性环氧树脂为上述方案所述的改性环氧树脂或上述方案所述制备方法制备的改性环氧树脂。
[0056]
以质量分数计,所述uv环氧树脂灌封胶包括多元醇19~46%,优选为25~40%,更优选为30~36%。在本发明中,所述多元醇优选包括二元醇、三元醇和超支化多元醇中的一种或多种。在本发明中,所述二元醇优选包括频哪醇;所述超支化多元醇优选包括聚己内酯多元醇;所述三元醇优选包括三羟甲基丙醇。在本发明中,多元醇的作用是uv固化后增韧和充当其分散溶剂。
[0057]
以质量分数计,所述uv环氧树脂灌封胶包括固化剂1~9%,优选为3~8%,更优选为4~6%。在本发明中,所述固化剂优选包括酸酐类固化剂、芳香基固化剂、有机酸固化剂和杂多酸固化剂中的一种或多种。在本发明中,所述酸酐类固化剂优选包括苯酮四羧酸二酐、四氢邻苯二甲酸酐、戊二酸酐和聚壬二酸酐中的一种或多种;所述有机酸固化剂优选包括己二酸、壬二酸、硬脂酸和月桂酸中的一种或多种;所述杂多酸固化剂优选包括钼磷杂多酸,钨磷杂多酸砷钨杂多酸和硅钨杂多酸中的一种或多种;所述芳香基固化剂优选包括二芳基碘鎓、三芳基硫鎓盐、芳香重氮盐和二氨基二苯甲基甲烷中的一种或多种。
[0058]
以质量分数计,所述uv环氧树脂灌封胶包括光敏剂1~9%,优选为3~8%,更优选为4~6%。在本发明中,所述光敏剂优选包括共轭联苯类有机物;所述共轭联苯类有机物优选包括二苯甲酮、硫杂蒽酮和安息香二甲醚中的一种或多种。在本发明中,光敏剂可以吸收诸如紫外线或可见光等具有更宽光谱范围的光波,然后将能量转换为光引发剂以促进聚合反应。
[0059]
以质量分数计,所述uv环氧树脂灌封胶包括填料0.5~3%,优选为优选为1~
2.5%,更优选为1.5~2%。在本发明中,所述填料优选包括无机填料和改性无机填料,所述无机填料优选包括氧化铝和/或氮化硼;所述改性无机填料优选包括hdpe填充滑石粉。
[0060]
以质量分数计,所述uv环氧树脂灌封胶优选包括光引发剂0.5~3%,优选为优选为1~2.5%,更优选为1.5~2%。在本发明中,所述光引发剂优选包括硼酸碘鎓盐。
[0061]
以质量分数计,所述uv环氧树脂灌封胶优选还包括稀释剂3~10%,优选为5~9%,更优选为6~8%。在本发明中,所述稀释剂优选包括丁基缩水甘油醚、新戊二醇缩水甘油醚、乙二醇、丁醇、异丙酮、甲苯和丁酮中的一种或多种。
[0062]
以质量分数计,所述uv环氧树脂灌封胶优选还包括促进剂0.5~5%,优选为1~4%,更优选为2~3%。在本发明中,所述促进剂优选包括咪唑、季磷盐和脲类衍生物中的一种或多种。在本发明中,所述咪唑优选包括2-甲基咪唑、1,3-二甲基咪唑氯盐和2-巯基苯丙咪唑锌盐中的一种或多种;所述季磷盐优选包括4-羧基丁基三苯基溴化鏻、氯化三丁基十四烷基磷和十烷基三丁基溴化鏻中的一种或多种;所述脲类衍生物优选包括乙酰脲、环己基脲和二苯脲中的一种或多种。
[0063]
以质量分数计,所述uv环氧树脂灌封胶优选还包括增韧剂0.5~2%,优选为0.8~1.6%,更优选为1~1.5%。在本发明中,所述增韧剂优选包括聚丁二烯橡胶、环氧化聚丁二烯、1,3丁二烯官能团的环氧树脂、乙丙橡胶和丁基橡胶中的一种或多种。
[0064]
以质量分数计,所述uv环氧树脂灌封胶包括上述方案所述的改性环氧树脂或上述方案所述制备方法制备的改性环氧树脂余量。
[0065]
本发明的uv环氧树脂灌封胶仅通过uv固化,就可达到较快uv固化速度和较高的固化深度;抗沉降性能好;uv固化后颜色无色透明、无气泡、表面平整、有光泽、硬度高、韧性高、抗弯折能力强、耐指划痕性能好、载带粘接性强;固化物耐酸碱性能好,防潮防水防油防尘性能佳,耐湿热和大气老化;具有良好的绝缘、抗压、粘接强度高等电气及物理特性。
[0066]
在本发明中,所述uv环氧树脂灌封胶的制备方法优选包括以下步骤:
[0067]
将多元醇、固化剂、光敏剂和改性环氧树脂进行第一混合,得到第一混合物;
[0068]
将所述第一混合物与填料进行第二混合,得到所述uv环氧树脂灌封胶。
[0069]
在本发明中,所述第一混合的温度优选为100~135℃。在本发明中,所述第一混合优选在搅拌条件下进行,本发明对所述第一混合的时间没有特殊的限定,混合均匀即可。
[0070]
在本发明中,所述将多元醇、固化剂、光敏剂和改性环氧树脂进行第一混合优选包括:将部分多元醇与固化剂、光敏剂、改性环氧树脂混合后再与剩余多元醇进行混合。分两次加多元醇有利于在第一次加入时增大未反应完全环氧树脂与非活性纳米离子的浓度,使未反应完全的环氧树脂与非活性纳米硅粉可以继续反应,提高反应速率,同时分两次加入更有利于体系混合均匀。
[0071]
第一混合后,本发明优选将所得混合产物过滤、冷却至室温,得到所述第一混合物。
[0072]
在本发明中,当所述uv环氧树脂灌封胶还含有光引发剂和/或促进剂时,本发明优选将所述光引发剂和/或促进剂优选与固化剂、光敏剂、改性环氧树脂一起进行混合。
[0073]
在本发明中,当所述uv环氧树脂灌封胶还含有稀释剂和/或增韧剂时,本发明优选将所述稀释剂和/或增韧剂优选同填料一起与第一混合物进行混合。
[0074]
为了进一步说明本发明,下面结合实施例和附图对本发明提供的一种改性环氧树
脂及其制备方法、uv环氧树脂灌封胶进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0075]
实施例1
[0076]
将300g聚合度为60的双酚a环氧树脂预热至80℃,在200r/min的搅拌速度下以0.1g/次的用量分200次加入粒径为90nm的非活性纳米硅粉,搅拌1h后升温至120℃;然后加入0.08gγ-氨基丙基三乙氧基硅烷偶联剂,在120℃的温度下以200r/min的搅拌速度搅拌2h发生第一缩合反应,再在200r/min的搅拌速度下冷却至室温后真空干燥并进行在200r/min的搅拌速度下进行第二缩合反应12h,得到改性环氧树脂;所述非活性纳米硅粉由液相急等法制备得到;所述改性环氧树脂中非活性纳米硅粉的接枝量为0.21%;
[0077]
将40g频哪醇、160g聚己内酯多元醇、20g二芳基碘鎓、30g二苯甲酮和630g实施例1的改性环氧树脂在100℃的温度搅拌6h,再加入5g氧化铝,混合均匀后,然后再105℃真空下搅拌2.5h,即得无色透明的uv环氧树脂灌封胶;
[0078]
对实施例1中的双酚a环氧树脂和实施例1制得的改性环氧树脂进行红外分析,结果如图5所示。其中蓝线是双酚a环氧树脂红外谱图,红色是环氧树脂和非活性硅粉预处理后的红外谱图。由图5可知,蓝线3600-3800cm-1
是双酚a环氧树脂-oh的伸缩振动峰,1500cm-1
和1200cm-1
是-c-o-伸缩振动峰,1000cm-1
和800cm-1
是-c-o-弯曲振动峰。红线是环氧树脂与活性硅粉预处理后体系红外谱图,3600-3800cm-1-oh伸缩振动峰消失,1000-1500cm-1
清晰分明的-c-o-伸缩振动峰变成一个大包。原因是双酚a环氧树脂分子上的羟基与非活性硅粉表面羟基反应所致。同时,由蓝线和红线纳米硅粉键合前后树脂体系ir叠加后进行质量检测分析可知,质量检测相关度仅为0.249(小于0.85为两种不同物质),也证实了预处理后双酚a环氧树脂与非活性硅粉表面的羟基发生了反应。
[0079]
在600s/1的剪切速率下,实施例1制备得到的改性环氧树脂的粘度为7000cps;在600s/1的剪切速率下,实施例1的双酚a环氧树脂粘度为5000cps。均匀分散的纳米粒子/环氧树脂共混体系的流变性能由粒子的微观结构、粒子及粒子与树脂间相互作用共同决定。环氧树脂和非活性纳米硅粉形成的化学键使改性环氧树脂具有强相互作用,所以不易剪切变稀。
[0080]
实施例1的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为90s,固化深度0.30mm,邵氏硬度为85;拉伸强度为66.40mpa;弯曲强度为133.2mpa;冲击强度为13.40kj/mm2;固化后无表面指划痕;对载带的粘接性失效模式良好,弯折测试表现韧性较高。
[0081]
实施例2
[0082]
将320g聚合度为100的双酚f环氧树脂预热至90℃,再加入200g聚合度为60的双酚s环氧树脂,在200r/min的搅拌速度下以0.1g/次的用量分100次加入粒径为100nm的非活性纳米硅粉,搅拌1h后升温至110℃;然后加入0.15g苯胺甲基三乙氧基硅烷偶联剂,在120℃的温度下以200r/min的搅拌速度搅拌2h发生第一缩合反应,再在200r/min的搅拌速度下冷却至室温后真空干燥并在200r/min的搅拌速度下进行第二缩合反应12h,得到改性环氧树脂;所述非活性纳米硅粉由化学还原法制备得到;所述改性环氧树脂中非活性纳米硅粉的接枝量为0.24wt%;
[0083]
将150g聚己内酯多元醇,25g三芳基硫翁盐,25g硫杂蒽酮和300g实施例2得到改性
环氧树脂混合,并加热升温至135℃,再加入20g聚己内酯多元醇过滤,冷却至室温加入25g丁基缩水甘油醚溶剂,10g填充hdpe的滑石粉,然后在105℃的真空条件下搅拌2.5h,即得无色透明uv环氧树脂灌封胶。
[0084]
实施例2的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为85s,固化深度0.25mm,邵氏硬度为90;固化后无表面指划痕;对载带的粘接性失效模式良好,弯折测试表现韧性较高。
[0085]
实施例3
[0086]
将100g聚合度为140的双酚a环氧树脂预热至90℃,再加入500g聚合度为40的双酚s环氧树脂,在200r/min的搅拌速度下以0.1g/次的用量分150次加入粒径为8nm的非活性纳米硅粉,搅拌1h后升温至80℃;然后加入0.2g乙烯基三乙氧基硅烷偶联剂,在120℃的温度下以200r/min的搅拌速度搅拌2h发生第一缩合反应,再在200r/min的搅拌速度下冷却至室温后真空干燥并在200r/min的搅拌速度下进行第二缩合反应12h,得到改性环氧树脂;所述非活性纳米硅粉由气相沉积法制备得到;所述改性环氧树脂中非活性纳米硅粉的接枝量为0.23wt%;
[0087]
将100g聚己内酯多元醇、90g三羟甲基丙醇,30g芳香重氮盐,30g安息香二甲醚和180g实施例3得到改性环氧树脂混合,并加热升温至135℃,再加入10g三羟甲基丙醇,过滤,冷却至室温加入增韧剂聚丁二烯橡胶8g和hdpe填充滑石粉12g,在80℃的真空条件下搅拌4h,即得改性环氧树脂灌封胶。
[0088]
实施例3的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为90s,固化深度0.20mm,邵氏硬度为85;固化后无表面指划痕;对载带的粘接性失效模式良好,弯折测试表现韧性较高。
[0089]
实施例4
[0090]
将340g聚合度为150的双酚a环氧树脂预热至90℃,再加入200g聚合度为80的双酚f环氧树脂,在200r/min的搅拌速度下以0.1g/次的用量分100次加入粒径为100nm的非活性纳米埃洛石,搅拌1h后升温至110℃;然后加入0.15g苯胺甲基三乙氧基硅烷偶联剂,在120℃的温度下以200r/min的搅拌速度搅拌2h发生第一缩合反应,再在200r/min的搅拌速度下冷却至室温后真空干燥并在200r/min的搅拌速度下进行第二缩合反应12h,得到改性环氧树脂;所述纳米埃洛石由化学还原法制备得到;所述改性环氧树脂中非活性纳米埃洛石的接枝量为0.21wt%;
[0091]
将150g聚己内酯多元醇,25g三芳基硫翁盐,25g硫杂蒽酮和300g实施例4得到改性环氧树脂混合,并加热升温至135℃,再加入20g聚内酯多元醇过滤,冷却至室温加入25g丁基缩水甘油醚溶剂,10g填充hdpe的滑石粉,然后在105℃的真空条件下搅拌2.5h,即得无色透明uv环氧树脂灌封胶。
[0092]
实施例4的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为90s,固化深度0.23mm,邵氏硬度为88;固化后无表面指划痕;对载带的粘接性失效模式良好,弯折测试表现韧性较高。室温下的沉降时间是25天。
[0093]
对比例1
[0094]
将聚合度为60的双酚a环氧树脂,粒径为90nm的非活性纳米硅粉(液相急冷法制得)、0.8gγ-氨基丙基三乙氧基硅烷偶联剂、40g频哪醇、160g聚己内酯多元醇、20g二芳基
碘鎓、30g二苯甲酮和630g聚合度为60的双酚a环氧树脂与粒径为90nm的非活性纳米硅粉的混合物(其中聚合度为60的双酚a环氧树脂600g,粒径为90nm的非活性纳米硅粉30g)、5g氧化铝,混合搅拌15小时后,即得无色透明的uv环氧树脂灌封胶。
[0095]
对比例1的uv环氧树脂灌封胶在能量为120mj/cm3的光照下固化时间为230s,固化深度0.13mm,邵氏硬度为50;拉伸强度为40.6mpa;弯曲强度为60.30mpa;冲击强度为6.30kj/mm2。
[0096]
在同等加入量的情况下,与对比例1相比,实施例1~3的uv固化时间减少了50%~70%,固化深度增加了60%~80%,同时无表面指划痕,邵氏硬度由50升高到85~90。
[0097]
对比例2
[0098]
与实施例2不同的仅是:将聚合度为100的双酚f环氧树脂替换为聚合度为100的环氧树脂epp175;将聚合度为60的双酚s环氧树脂替换为聚合度为200的环氧树脂epp180。
[0099]
对比例3
[0100]
与实施例3不同的仅是:将聚合度为140的双酚a环氧树脂替换为聚合度为10的双酚a环氧树脂;将聚合度为40的双酚s环氧树脂替换为聚合度为10的双酚s环氧树脂;非活性纳米硅粉的粒径为1.2μm;非活性纳米硅粉由机械球磨法制备得到。
[0101]
按照《astm d6930-10乳化沥青的沉降和储存稳定性的标准试验方法》测定实施例1~3,对比例1~3的uv环氧树脂灌封胶的沉降时间。结果为:实施例1~3的uv环氧树脂灌封胶室温下的沉降时间均为25天,对比例1的uv环氧树脂灌封胶室温下的沉降时间为21天,对比例2的uv环氧树脂灌封胶室温下的沉降时间为20天,对比例3的uv环氧树脂灌封胶室温下的沉降时间为20天。
[0102]
将对比例1~2、实施例1的uv环氧树脂灌封胶按照相同的条件(uv光照能量120mj/cm3,固化时间90s)分别固化在载带后再从载带上剥离下来,用扫描电镜拍摄剥离断面,结果如图2~4所示。
[0103]
由图2~4可知,纯环氧树脂的断裂面表面相对光滑,属于脆性断裂模式;硅粉混合后树脂断面与纯氧树脂相比相对粗糙,微裂纹变多;改性环氧树脂的断面粗糙程度最大,微裂纹更多。改性之后,纳米非活性硅粉与双酚a环氧树脂既通过物理作用又通过化学作用结合在一起,物理作用是指它们之间存在的范德华力,即存在于高分子链之间的纳米粒子可以改变链间的作用力;化学作用是纳米粒子由于小尺寸使其表面活性点和大分子之间可以形成化学键的结合。预处理后的纳米非活性硅粉与双酚a环氧树脂具有纳米粒子的表面效应、体积效应、量子尺寸效应和宏观量子隧道效应等综合作用。当uv光照射时,当树脂处于自由状态下,反应过程中单体分子间距离转化为共价键距离,宏观上表现为体积收缩。uv光照射初期,uv光的一部分能量消耗用于树脂分子之间转化为共价键距离。除此之外,uv光照射初期,由于热量增加会引起树脂膨胀,这也会影响uv固化的时间。将纳米非活性硅粉对双酚a环氧树脂改性后,纳米非活性硅粉与双酚a环氧树脂通过化学键键合,缩短了uv固化时间。
[0104]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1